

 и/или концентрации гемоглобмна (N мужчины 160±20г/л; ж…")
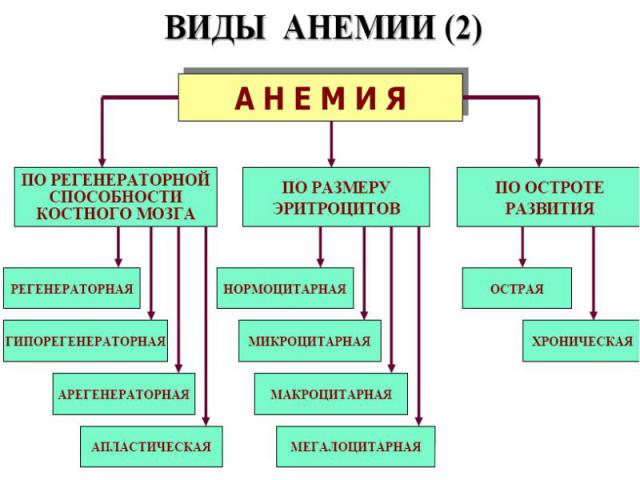
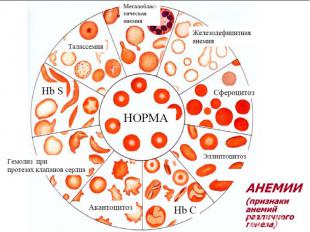
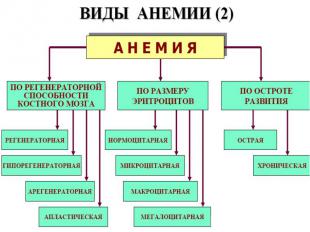
 ВИДЫ АНЕМИИ (3) По цветовому показателю Нормохромные (0,8-1,0) Гиперхромные (˃1,0) Гипохромные (˂0,8) По конц-ции Нв степень анемии: 80-100 г/л легкая 60-80 г/л средняя ˂60 г/л тяжелая")
 хронические, острые Хроническая ПГА. Причины: повторяющиеся кровотечения из внутренних органов. Явл-ся частным вариантом железо-дефицитных анемий. Гематолог.синдром: гипорегенераторная, гипохромная, микроцитарная. Ос…")
; Кровопотеря средней тяжести -25-35%; Тяжелая форма анемии развивается…")
. Ht, Э, Нb - норма. «Скрытая анемия» - нормохромная, нормоцитарная. Клинические проявления характерны для коллапса: ↓ АД, бле…")

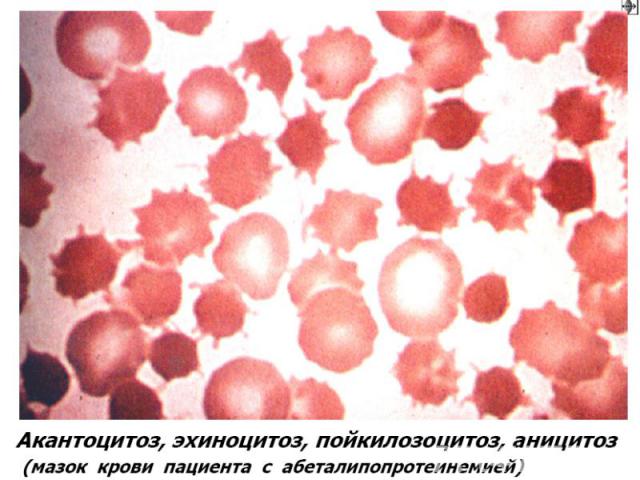
 Патогенез – врожденные дефекты клеточной мембраны. Нар-ие синтеза белков (спектрин, анкерин и др.) Семейный наследственный микросфероцитоз (болезнь Минковского-Шоффара). Наследуется аутосомно-доминантно. Мембранопатия…")
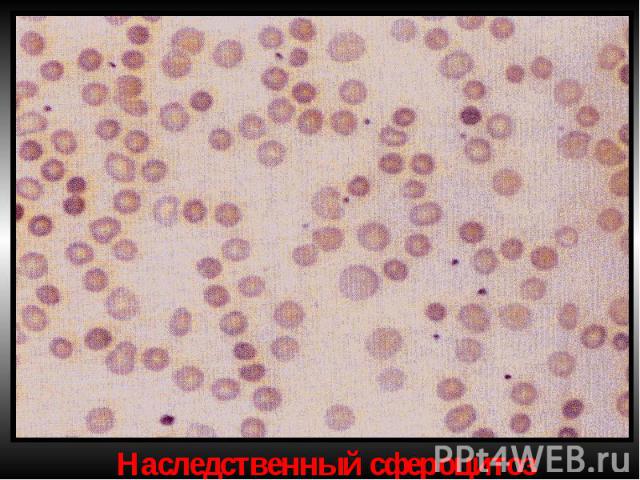
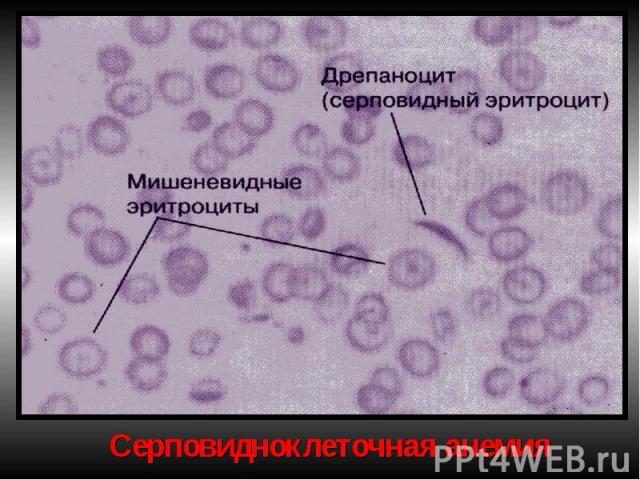
 на другую (валин). В основе образования серповидных клеток …")
 Талассемия (греч. talasia — море) ⇒ дефицит HвA без качественных нарушений его глобиновых цепей. Патология генов-регуляторов ⇒ в эмбриогенезе не происходит норм. переключения синтеза глобиновых цепей, обр-ся аномальный…")


 АТ. Вызванные холодовыми (Igм) АТ. Анемия с холо…")

 под действием агентов физ., хим., биол.природы. Патогенез – нар-ие пролиферации и/или гибель стволовых гемопоэтич. клеток→анемия. Проявле…")
 дефицитные анемии и фолиево-дефицитные анемии (мегалобластические). Дефицит В…")
 дефицитных анемий: Анемия Аддисона-Бирмера. Аутоиммунное заболевание. Цитотоксический тип поражения слизистой желудка с п…")
 «синий» КМ, а также мегалокариоциты. Нарушается дифференцировка и других клеток миелоидного ряда. В периферической крови значит…")
 и пул хранения (гемосидерин и ферритин). Ферритин – комплекс железа с белком (во многих тканях, много в макрофагах). Часть феррити…")
, пойкилоцитоз. Изменения органов и тканей:…")
. Эритропоэтическая стимуляция КМ несовершенна, особенно у недоноше…")

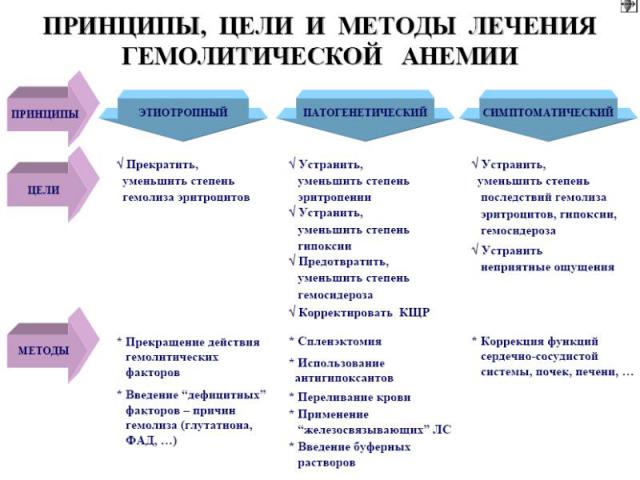
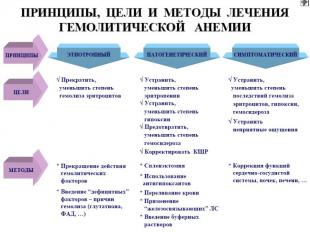
; устранение гипоксии; предотвращение гемосидероза; коррекция КЩР; устранение последствий гипоксии.")

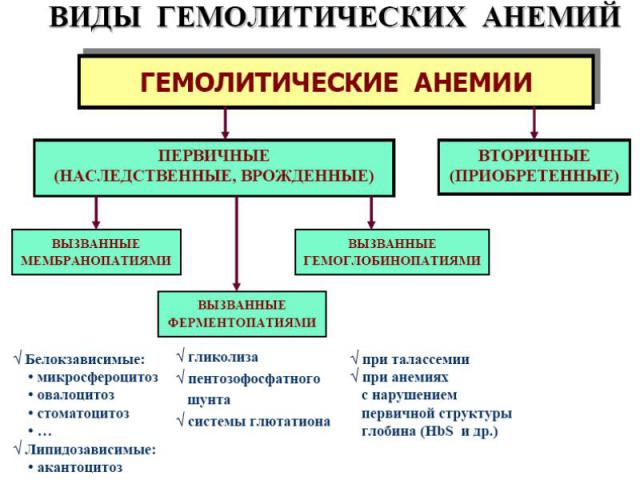
Презентация на тему: Патофизиология системы крови нарушения системы эритроцитов

ГОУ ВПО «Красноярский государственный медицинский университет имени проф. В.Ф. Войно-Ясенецкого Министерства здравоохранения и социального развития Российской Федерации» Лекция ПАТОФИЗИОЛОГИЯ СИСТЕМЫ КРОВИ НАРУШЕНИЯ СИСТЕМЫ ЭРИТРОЦИТОВ Доцент кафедры патологической физиологии с курсом клинической патофизиологии Фефелова Ю.А.

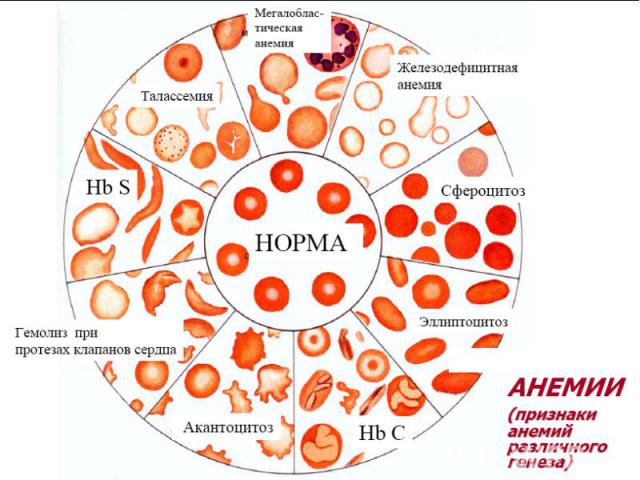
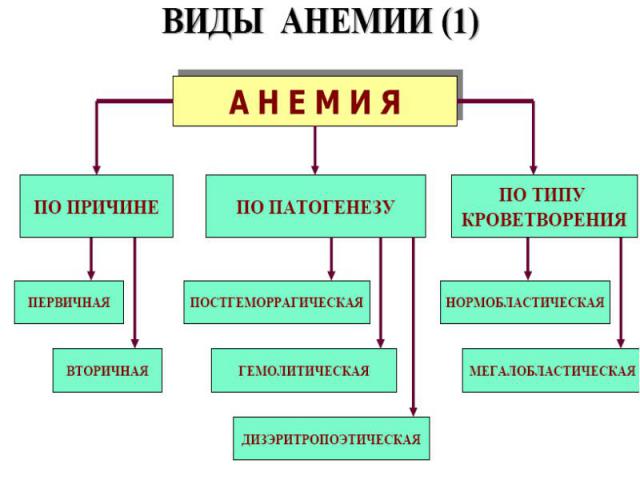
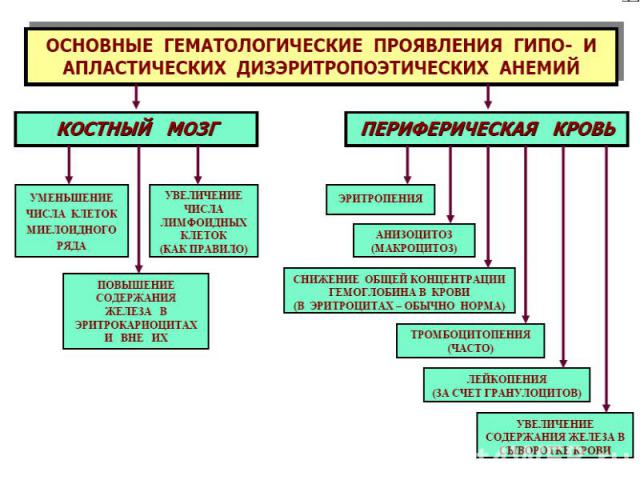
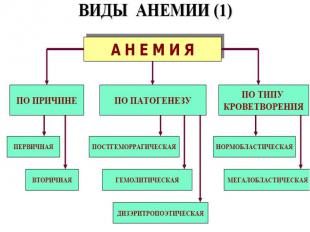
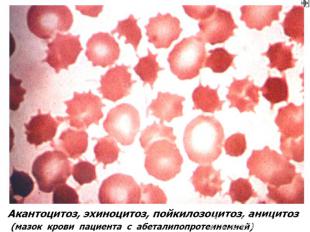
2 формы патологии эритроцитарной системы: Анемии Эритроцитозы Анемия – клинико-гематологический синдром, характеризующийся ↓ кол-ва эритроцитов (N мужчины 5,5±0,9·1012/л; женщины 4,8±0,6·1012/л) и/или концентрации гемоглобмна (N мужчины 160±20г/л; женщины 140±20г/л) в ед. объема крови. Главная патофизиологическая суть анемии ↓ кислородной емкости крови, приводящей к гипоксии гемического типа. С гипоксией связаны основные клинические симптомы и расстройства жизнедеятельности у больных анемией. Пойкилоцитоз – состояние, при котором встречаются эритроциты необычной формы. Анизоцитоз – различие в величине клеток в одной ткани или клеточной популяции из-за нарушения функции КМ.
 ВИДЫ АНЕМИИ (3) По цветовому показателю Нормохромные (0,8-1,0) Г")
ВИДЫ АНЕМИИ (3) ВИДЫ АНЕМИИ (3) По цветовому показателю Нормохромные (0,8-1,0) Гиперхромные (˃1,0) Гипохромные (˂0,8) По конц-ции Нв степень анемии: 80-100 г/л легкая 60-80 г/л средняя ˂60 г/л тяжелая
 хронические, острые Хроническая ПГА. Причины: п")
Постгеморрагические анемии (ПГА) хронические, острые Хроническая ПГА. Причины: повторяющиеся кровотечения из внутренних органов. Явл-ся частным вариантом железо-дефицитных анемий. Гематолог.синдром: гипорегенераторная, гипохромная, микроцитарная. Острая ПГА. Развиваются в результате массивной кровопотери (внешние травмы, кровотечения из внутренних органов). Ведущее патогенетическое звено ↓ ОЦК → гипоксия → нар-ия КОС, дисбаланс ионов.

Выраженность анемии от степени кровопотери При потере 15-25% ОЦК развивается легкая постгеморрагическая анемия (для детей характерна более высокая чувствительность к кровопотере); Кровопотеря средней тяжести -25-35%; Тяжелая форма анемии развивается при кровопотере 35-50% объема крови; Более 50% кровопотери летально.

Фазы течения острой ПГА Рефлекторная фаза компенсации. Картина крови не отличается от нормы (нормоцитемическая гиповолемия). Ht, Э, Нb - норма. «Скрытая анемия» - нормохромная, нормоцитарная. Клинические проявления характерны для коллапса: ↓ АД, бледность, тахикардия, тахипноэ. Гидремическая фаза (следствие раздражения волюморецепторов)► полидипсия ► ↑поступление жидкости извне, тканевая жидкость переходит в сосуды, спазм сосудов почек , ↓ диурез, активация РААС, задерживается Na и вода). Выброс Эр. из депо. После восстановления ОЦК появляются гематологические симптомы. Эритроциты крови и выброшенные из депо содержат нормальное количество гемоглобина (анемия нормохромная). Гипоксия стимулирует выделение эритропоэтинов почками►↑ эритропоэз. Фаза костно-мозговой компенсации. В крови ретикулоцитов до 30-40% (гиперрегенераторная анемия). ЦП <0,85 (гипохромная анемия), скорость синтеза Нb отстает от темпа пролиферации клеток. В костном мозге - признаки интенсификации эритропоэза — ↑ кличество эритробластов и ретикулоцитов. Длится до 14 дней.

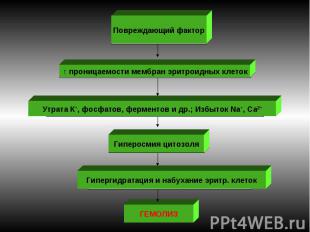
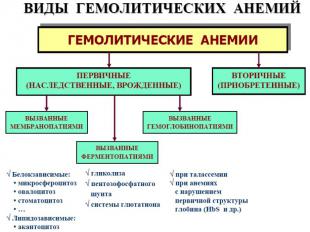
Гемолитические анемии ↓ ср. продолжительности жизни эритроцитов в результате преобладания процесса гемолиза над их продукцией. Причины приобретенных ГА - действие: факторов физического характера. Искусственные клапаны сердца, множественные протезы сосудов, «маршевая» гемоглобинурия, при тромбообразовании в мелких сосудах (сдавливается мембрана эритроцитов или «разрезается» нитями фибрина) и т.д.; химических факторов — это «гемолитические яды». Соединения свинца, цинка, фосфора, нитробензол, сульфаниламиды, фенацетин; биологических — грибной, змеиный, пчелиный яды, экзо-и эндотоксины бактерий, продукты метаболизма паразитов (малярийный плазмодий), аутоантитела на эритроциты (наибольший удельный вес среди причин приобретенных ГА).
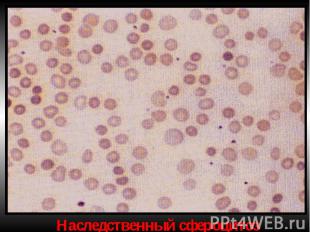
 Патогенез – врожденные дефекты клеточной мембран")
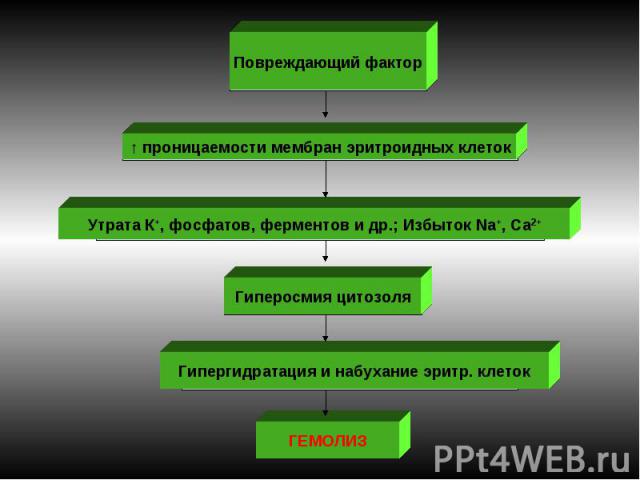
Мембранопатии (эритроцитопатии) Патогенез – врожденные дефекты клеточной мембраны. Нар-ие синтеза белков (спектрин, анкерин и др.) Семейный наследственный микросфероцитоз (болезнь Минковского-Шоффара). Наследуется аутосомно-доминантно. Мембранопатия обусловлена значительным ↓ содержания белка спектрина, нарушением связывания его с другими белками мембран. Структура самого спектрина при микросфероцитозе изменена. Все это обусловливает ↑ проницаемости мембраны эритроцитов для ионов Nа+,Са²+ и накопление их избытка, а также жидкости в гиалоплазме. Гипергидратированные эритроциты приобретают сферическую форму. Это ↓ пластичность мембран эритроцитов, их способность к деформации в микрососудах. Проходя через селезеночные синусы, они не могут деформироваться, теряют часть поверхности и превращаются в сфероциты малого размера, резко ↓ их продолжительность жизни (до 6-8 суток). Характеризуется длительным латентным течением часто с одним симптомом желтухи с микросфероцитозом. Провоцируют обострение переохлаждение, переутомление, инфекции. В остром периоде - спленомегалия и желтуха с уробилирубинемией и уробилинурией, а также ↑t°. Часто возникают трофические язвы (следствие микротромбов при гемолизе).

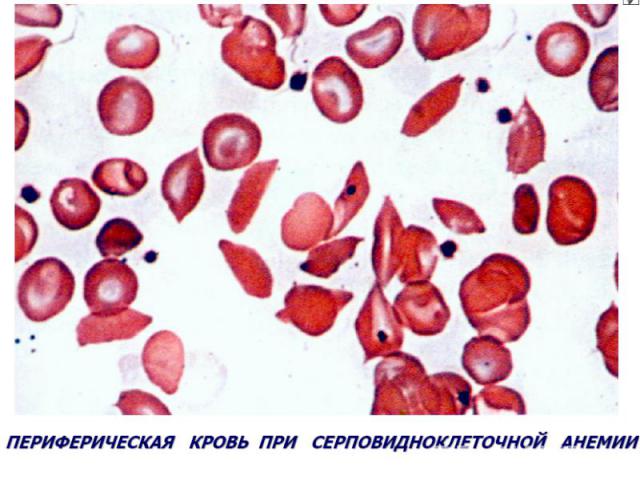
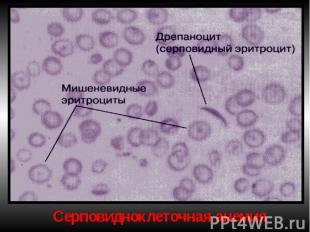
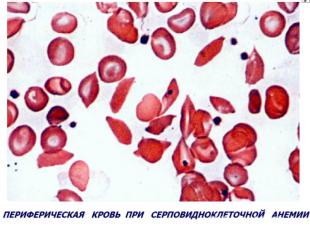
Гемоглобинопатии Серповидно-клеточная анемия. Патогенез — генетический дефект в структурном гене ⇒ нарушается структура β-цепей глобина в связи с заменой одной аминокислоты (чаще глютамина) на другую (валин). В основе образования серповидных клеток лежит свойство HbS полимеризоваться при переходе в восстановленную форму. Образующиеся полимеры (длинные нити) группируются в тактоиды, они изменяют форму и вид эритроцитов. Полимеризация восстановленного HbS связана с его низкой растворимостью (в 100 раз ↓ растворимости HbA). Образование тактоидов зависит от концентрации HbS в эритроците и парциального давления кислорода в крови. ↓Нв до 30-50 г/л, ретикулоцитоз, ↑ сод-ия железа в сыворотке, гипохромия.
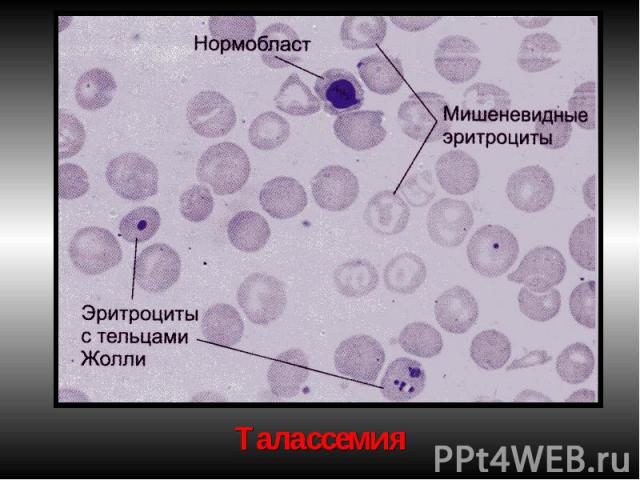
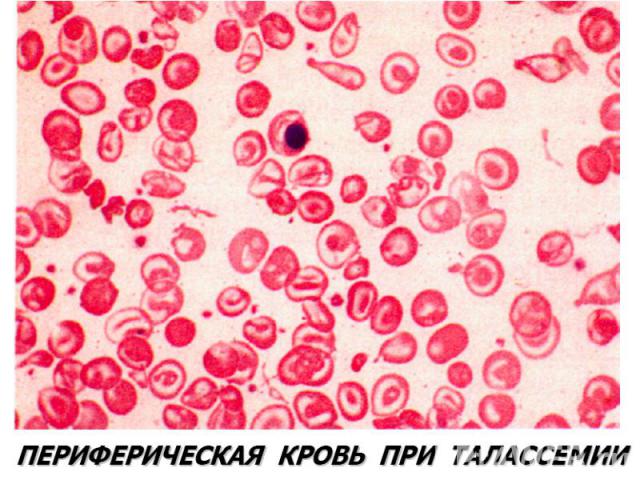
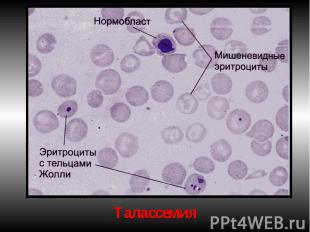
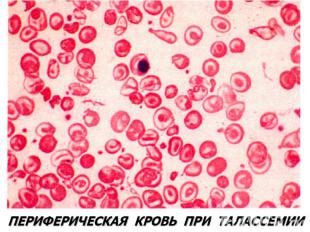
 Талассемия (греч. talasia — море) ⇒ дефицит HвA б")
Талассемии (б. Кули-Кули) Талассемия (греч. talasia — море) ⇒ дефицит HвA без качественных нарушений его глобиновых цепей. Патология генов-регуляторов ⇒ в эмбриогенезе не происходит норм. переключения синтеза глобиновых цепей, обр-ся аномальный Нв в ущерб синтезу HbA. Две группы: α-талассемия и β-талассемия. При α-талассемии - частичная или полная делеция α-глобиновых генов-регуляторов → ↓ синтез этих цепей. Их недостаток комп-ся в эмбр. периоде избыточным синтезом ץ-глоб. цепей (Нb Bart's). После рождения недостаток α-глобиновых цепей восполняется синтезом β-глобиновых цепей (HbH). (HbBart's и HbH - маркеры α-талассемии). Клиническое проявление α-талассемии — гипоксия из-за высокого сродства HbBart's и HbH к О2. Анемия усугубляется ↑ разрушением эритроцитов в селезенке.

β-талассемия. β-талассемия. Β0 β+ талассемии. Патогенез: 2 механизма: -Нв синтезируется меньше, гипохромия. -↓жизни эритроцитов из-за несбалансированного синтеза цепей Нв. Итог: неэффективность эритропоэза.

Энзимопатии Наиболее часто обнаруживают следующие дефекты: дефекты активности ферментов гликолиза: пируваткиназы, гексокиназы, фосфофруктокиназы и др. В эритроцитах ведущим путем ресинтеза АТФ является гликолиз. →→ недостаток энергии АТФ обусловливает нарушение трансмембранного переноса ионов. Развивается их дисбаланс ► гипергидратация и набухание эритроцитов; ↓ активности энзимов пентозофосфатного цикла. В ходе его реализации образуется восстановленная форма НАДФ, использующаяся для восстановления глютатиона. Восстановленный глютатион — компонент антиоксидантной системы эритроцитов► при таких энзимопатиях имеет место разрушение липопротеидных комплексов мембран; дефицит ферментов системы глютатиона (глютатионсинтетазы, глютатионредуктазы и т.д.). Последствия активация липоперекисных реакций в эритроцитах, нарушения целостности их мембраны. Пример: гемолитическая анемия, связанная с недостаточностью в эритроцитах Г-6-ФД. Гемолиз провоцируется приемом лекарственных препаратов (сульфаниламиды, антипиретики, анальгетики и др). При ↓ Г-6-ФД эритроциты быстро утрачивают минимум имеющегося восстановленного глутатиона и быстро стареют Анемия: нормохромная, нормоцитарная, регенераторная.

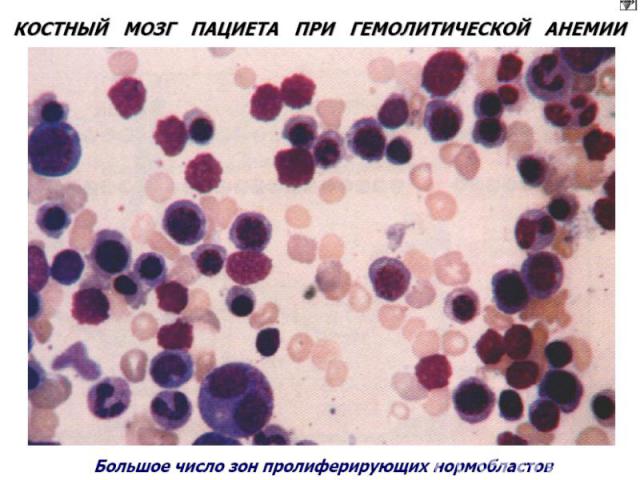
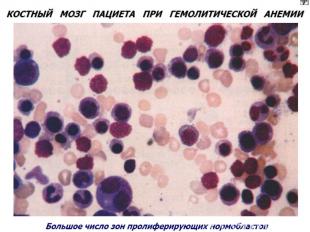
Приобретенные гемолитические анемии Генез различен. Анемии этой группы объединяет внутрисосудистый гемолиз. По мех-му разрушения эритроцитов: иммунные и неиммунные. Иммунные. Вызванные тепловыми (IgG) АТ. Вызванные холодовыми (Igм) АТ. Анемия с холодовыми (IgG) гемолизинами. Анемии неиммунного генеза. Причина: инфекц.заболевания, малярия, укусы змей, ожоговая болезнь и др. ---------------------------------------------------------------------------------------- Картина крови при гемолит.анемиях: эритропения, полихроматофилия, пойкилоцитоз, ретикулоцитоз, ↑непрямого билирубина. В КМ: ↑клеток эритроидного ростка.

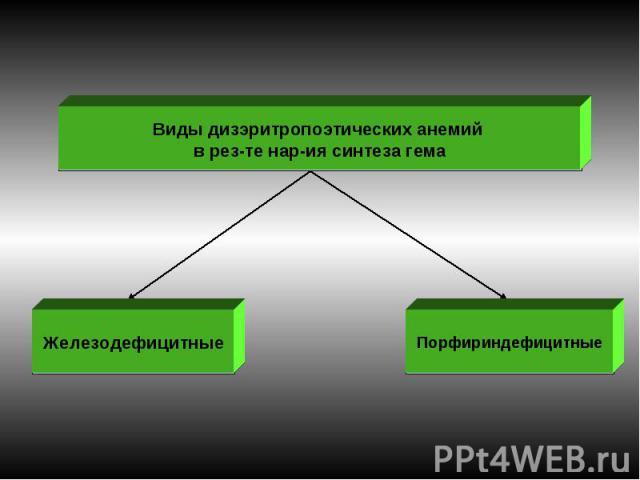
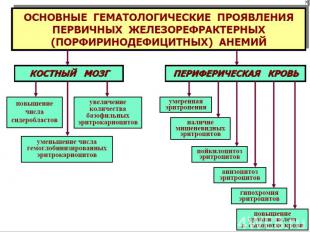
ДИЗЭРИТРОПОЭТИЧЕСКИЕ АНЕМИИ 1. Обусловленные нарушением эритропоэза в связи с преимущественным повреждением стволовых клеток: гипо- и апластические анемии. 2. Обусловленные нарушением эритропоэза в связи с преимущественным повреждением клеток- предшественников миелопоэза и/ или эритропоэтинчувствительных клеток: вследствие нарушения синтеза нуклеиновых кислот эритроплазмоцитов (мегалобластические): витамин В12 —(фолиево) дефицитные анемии; фолиеводефицитные анемии. в результате нарушения синтеза гема: железодефицитная анемия; порфириндефицитная; вследствие нарушения синтеза глобина: талассемии; при нар-ии первичной структуры цепей глобина (серп.-клеточ. ан.). При нар-ии регуляции деления и созревания эритроид.клеток.

1.Гипо- апластические анемии Являются синдромами др.пат. процессов. По происхождению: Приобретенные (вторичные) под действием агентов физ., хим., биол.природы. Патогенез – нар-ие пролиферации и/или гибель стволовых гемопоэтич. клеток→анемия. Проявления – гипоксия, кровотечения и кровоизлияния (из-за ↓сверт.крови), инфекции. Наследуемые (первичные). Анемия Фанкони. (Аутосомно-рецессивно). Патогенез – нар-ие процессов репарации ДНК стволовых клеток, ↑мутабельности. В целом не ясен, предположительно – иммунолог.супрессия, либо аномалии стволовых клеток. Проявления: гипоксия, тромбоцитопения, лейкопения (инфекции).

2. Обусловленные нарушением эритропоэза в связи с преимущественным повреждением клеток- предшественников миелопоэза и/или эритропоэтинчувствительных клеток. В12 —(фолиево) дефицитные анемии и фолиево-дефицитные анемии (мегалобластические). Дефицит В12 и фолиевой к-ты. Нар-ся синтез ДНК. В12 (кобаламин) и фолаты – коэнзимы в процессе синтеза ДНК. Их недостаток →дефект созревания ядра, нар-ие процессов деления. Асинхронность созревания цитоплазмы и ядра. Аномальность кроветворения. Для всасывания В12 необходим ф.Кастла. 2 формы В12: метилкобаламин (с его помощью фолиевая к-та превращается в тетрагидрофолиевую; при его отсутствии не обр-ся предшеств.ДНК, процесс обрывается, торможение наз-ся «капкан метилфолата»). аденозинкобаламин – дефицит ведет к накоплению метилмалоната и пропионата → избыток аномальных ЖК в липидах нервных структур → неврологическая симптоматика.

Клиническая триада при дефиците В12: Мегалобластическая анемия; Глоссит Хантера; Фуникулярный миелоз. Патогенез В12 — (фолиево) дефицитных анемий: Анемия Аддисона-Бирмера. Аутоиммунное заболевание. Цитотоксический тип поражения слизистой желудка с потерей париетальных клеток его фундального отдела. АТ 3 типов: I тип - блокирующие (блокируют связывание В12 с ф.Кастла), II тип – нар-ие связывания комплекса с рецептором, III тип – против микроворсинок париетальных клеток. Проявления мегалобласт.анемий: Общие для В12 (фолиево) и анемии при дефиците фолатов: связаны с нарушением синтеза ДНК (изменения в КМ, крови, ЖКТ). Различия: фолатная недостаточность не связана с потерей ф-ии аденозин-кобаламина в синтезе ЖК и расстройствами в периферич. и ЦНС.

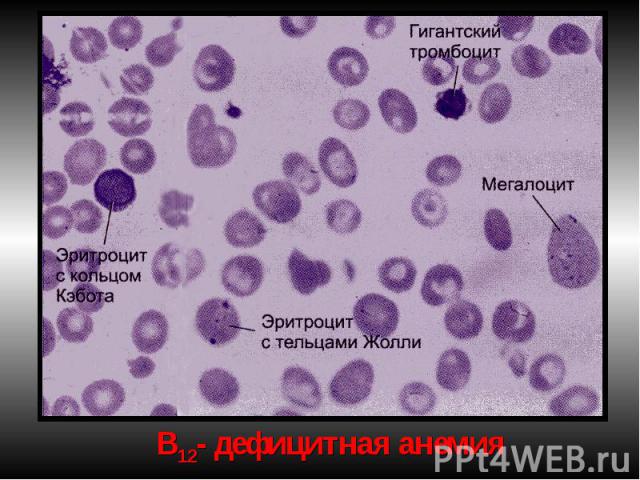
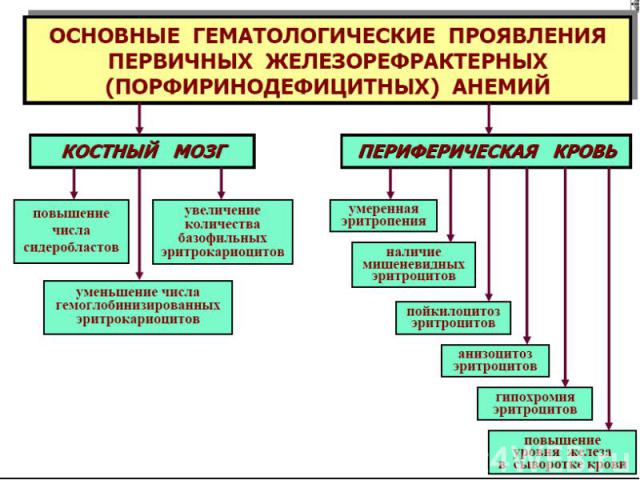
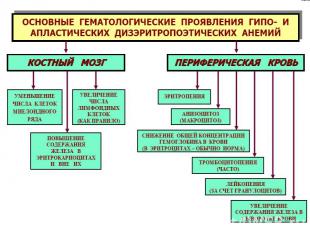
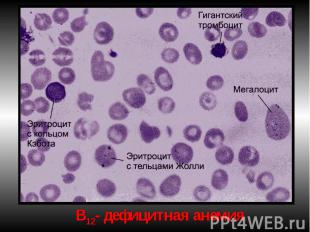
Проявления мегалобласт.анемий: Проявления мегалобласт.анемий: В костном мозге обнаруживаются мегалобласты (d > 15 мкм) «синий» КМ, а также мегалокариоциты. Нарушается дифференцировка и других клеток миелоидного ряда. В периферической крови значительно ↓ число эритроцитов, иногда до 0,7-0,8 х10¹²/л. Они большего размера, овальной формы, без центрального просветления. Встречаются мегалобласты. В эритроцитах - остатки ядерного вещества (тельца Жолли) и нуклеолеммы (кольца Кебота). Характерны анизоцитоз и пойкилоцитоз. ЦП=1,1–1,3. Нb в крови существенно ↓ (из-за↓числа эритроцитов). Количество ретикулоцитов чаще ↓, как правило, наблюдается лейкопения (за счет нейтрофилов). В связи с ↑гемолизом — билирубинемия. Мегалобластические анемии хар-ся как гиперхромные, макро- мегалоцитарные, гипо- арегенераторные.
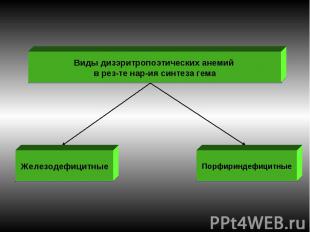
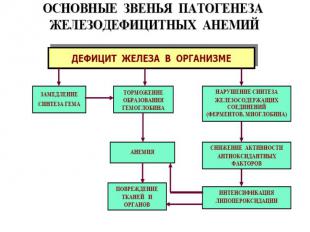

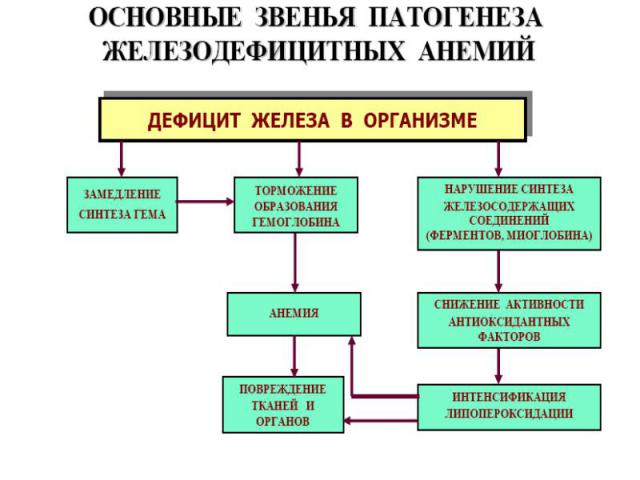
Железодефицитные анемии. ¾ всех анемий. Макс. – беременные и кормящие матери. Железо организма – функциональное (в Нв) и пул хранения (гемосидерин и ферритин). Ферритин – комплекс железа с белком (во многих тканях, много в макрофагах). Часть ферритина в макрофагах деградирует с образованием гемосидерина (гранулы). Индикатор дефицита или избытка железа – сывороточный ферритин. Осн.причина ЖДА – хроническая кровопотеря. Стадии дефицита железа: Скрытый дефицит. Латентный дефицит. Постоянный дефицит. Выраженный дефицит.

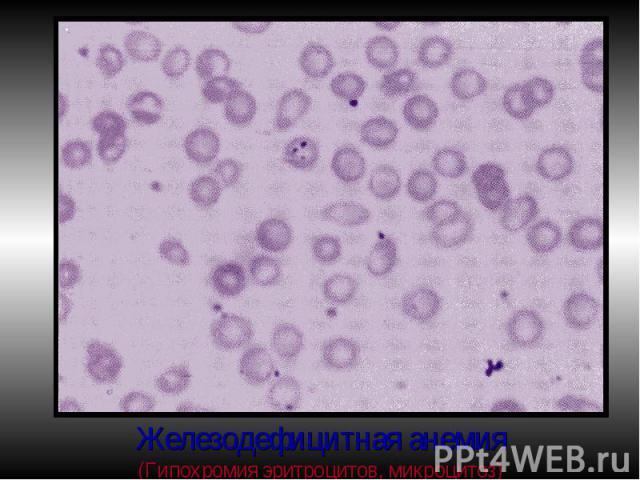
Проявления ЖДА: Проявления ЖДА: КМ: ↑ эритропоэза, исчезновение железа из макрофагов и ↓ индекса сидеробластов КМ. Периф.кровь: микроциты, гипохромия, анулоциты (Нв распологается в виде кольца на периферии), пойкилоцитоз. Изменения органов и тканей: глоссит, трещины в углах губ, отеки, извращение вкуса, хрупкость ногтей.
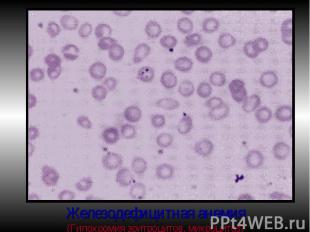

Для педиатр.ф-та. Для педиатр.ф-та. ЖДА болеют дети в возрасте до года. Кинетика эритрона отличается своей напряженностью (процессы естеств.убыли Эр. не восполняются их образованием). Эритропоэтическая стимуляция КМ несовершенна, особенно у недоношенных. Возникает физиологическая анемия детей 1-го года жизни. После первого года ослабевает пассивный естест.иммунитет, что ведет к инфекции →↓уровня трансферрина (ООФ), накопление железа в макрофагах (гемосидерин).

В молекуле гема железо связано с протопорфирином. Порфирины син-ся более всего в эритрокариоцитах и печени. Порфирины входят в сос-в пероксидаз, цитохромов, Нb, миоглобина.

Принципы и методы терапии ДА устранение причины нарушения деления эритроцитов (прекращение действия повреждающих факторов, введение дефицитных факторов); устранение гипоксии; предотвращение гемосидероза; коррекция КЩР; устранение последствий гипоксии.












