







 офтальмоплегия слабость мимических мышц (амимия, гипомимия, поперечная улыбка, маскоподобное лицо, лагофталь…")

 у 85 % больных скелетной формой и у 10 % с глазной формой Биопсия мышц - не имеет существенного диагно…")
. Эффект наступает через 10-30 мин, достигает максимума через 2 часа и длится еще 2 часа. Всего действие калим…")

 назначают при генерализованной и бульбарной формах от 50 мг/сутки до 150-200 мг/сутки. Клинически эффект наступает через 1,5-3 мес от начала лечения и достигает максимума до 1 года. Плазмаферез - при остром прог…")
. Побочные эффекты: гипоальбуминэмия, гипотензия, нарушения гемостаза внутривенное введение иммуноглобулина (2 г/кг веса 2-5 дней) кортикостероиди (преднизолон в дозе 100 мг…")


 Семейный спастический паралич с ихтиозом и…")



 Торзионная дистония Двойной атетоз Хорея Гентингтона Болезнь Паркинсона (дрожательный паралич) Миоклон…")
 – хроническое прогрессирующее дегенеративное заболевани…")
 сосудистый паркинсонизм медикаментозный (резерпин, антидепрессанты, бутифероны, антипсихотики, фенотиаз…")
 Наследственные заб…")
 медленная походка (пропульсии) ІІ. Ригидность мышц феномен „зубчастого колеса” увеличение ригидности в моме…")
 І - незначительное снижение активности, что не отражается на трудоспособности и выполнении профессиональных обязанности ІІ – значительное снижение профессиональной и быт…")
 начало с изолированного акинетико-гипертонического синдрома (потеря синхронности движений рук во время ходьбы, микрография, нарушение коротких и быстрых движений рукой (чистка зубов, взбивание омл…")

 агонисты дофаминовых рецепторов: - эрголинового ряда (бромкриптин, роналин, парлодел) - неэрголинового ряда (проноран…")



 (болезнь Вестфаля-Вильсона-Коновалова) – одна из наиболее изученных наследственых форм экстрапирамидных заболеваний, связана с нарушением обмена церулоплазмина – белка плазмы крови, что содержит медь, и синтезирует…")
 изменения психики могут наблюдаться судорожные присту…")

 Патоморфологически в основе дегенеративные изменения в подкорковых ганглиях, субталамических ядрах и зубчатом ядре мозжечка, нарушение синтеза и обмена медиаторов в базальных ганглиях Тип наследо…")





: Спинальная атаксия Фридрайха наследственная мозжечковая атаксия Пьєра Мари Разные клинические формы оливо-понто-церебелярной дегенерации Болезнь Рефсума Б…")
")













 Парамиотония Эйленбурга Синдром Шварта-Джампела (хондродистрофическая миотония)")


Презентация на тему: НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ НЕРВНОЙ СИСТЕМЫ

Наследственные заболевания нервной системы

Наследственные болезни нервной системы делят: Болезни с преимущественным поражением нервно-мышечного синапса Болезни с преимущественным поражением пирамидной системы Болезни с преимущественным поражением экстрапирамидной системы Болезни с преимущественным поражением мозжечковой системы (наследственные атаксии)

Наследственные заболевания нервной системы делят: Болезни с преимущественным поражением нервно-мышечной системы: І. Прогрессирующие мышечные дистрофии ІІ. Амиотрофии как результат поражения периферического двигательного нейрона Наследственно-семейные миотонии: Наследственные нервно-мышечные заболевания с пароксизмальными состояниями

Наследсвенные заболевания с преимущественным поражением нервно-мышечного синапса: Миастения Миастеноподобные синдромы

Миастения Наиболее распространенное среди известных синаптических заболеваний человека Этиология: неизвестна Описаны семейные случаи миастении, хотя наследственный характер миастении не установлен В большинстве случаев у больных находят опухоль или гиперплазию вилочковой железы

Патогенез уменьшение числа холинэргических рецепторов конечной пластинки недостаточная чувствительность холинэргических рецепторов к АХ нарушение синтеза АХ как результат дефекта активности фермента дефицит ацетилхолинэстеразы врожденная недостаточность вторичных синаптических щелей дефицит АХ-рецепторов аномалии кинетики АХ-рецепторов (то есть аномалии взаимодействия молекулы АХ и рецептора

Клиника За характером течения выделяют: прогрессирующая форма стационарная форма миастенические эпизоды Клинические формы: глазная форма бульбарная скелетная генерализованная

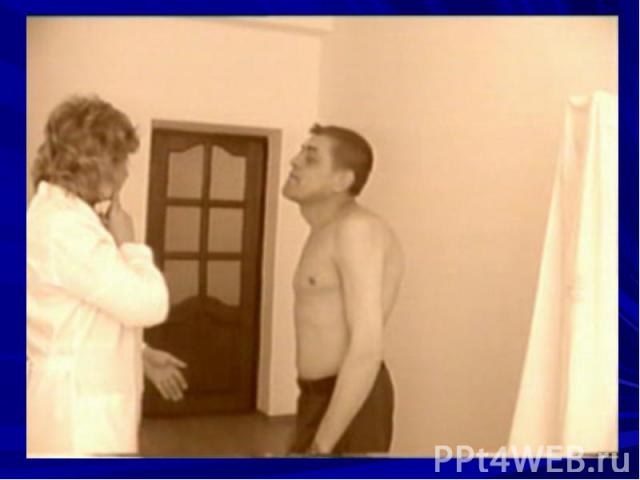
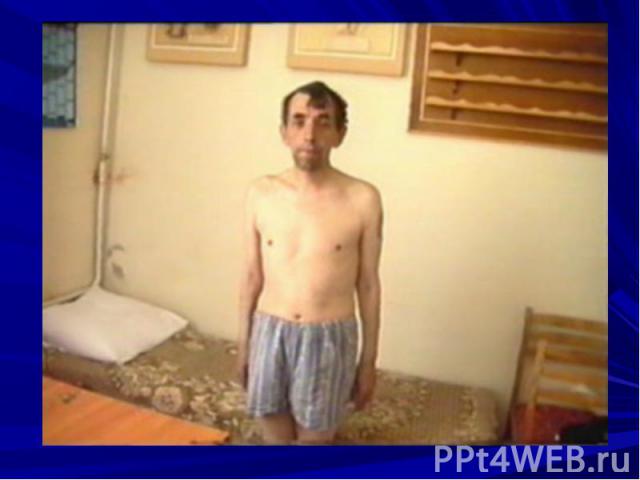
Характерные особенности: асимметричность поражения динамичность симптомов (в утренние часы больному лучше, вечером птоз и двоение нарастают) офтальмоплегия слабость мимических мышц (амимия, гипомимия, поперечная улыбка, маскоподобное лицо, лагофтальм) слабость жевальных мышц – во время еды больные утомляются слабость мышц глотки, гортани, языка нарушение функции языка нарушение дыхания нарушения функции конечностей слабость мышц шеи (свисание головы) слабость мышц туловища рефлексы в норме или истощаются мышечные атрофии (мышц языка, шеи, проксимальных отделов рук) чувствительные нарушения не наблюдаются тазовых нарушений не бывает

Пробы на мышечную утомляемость Для провокации птоза просят больного 30 сек смотреть либо вверх либо в сторону Дизартрию – читать текст Слабость жевальных мышц – произвести около 100 жевальных движений в закрытом рту Слабость мышц шеи – опустить голову и 1 минуту смотреть на свой пупок Слабость плечевого пояса – вытянуть руки вперед или в стороны и держать так 3 минуты Слабость ног – долго приседать. Феномен Уолкера: повторные сжимания и разгибания кисти вызывают слабость предплечий и даже усиление птоза 2. Прозериновая проба. Прозерин в дозе 1,5-3 мл 0,05 % вводят п/к. Через 20-40 мин – регресс всех симптомов. А через 2-3 часа – все возвращается на свои места. При глазной форме проба малоинформативна

Диагностика ЭМГ – миастеническая реакция, миастеническое истощение (прогрессирующее снижение амплитуды М-ответов больше, чем на 10-15 %. Проба (+) у 85 % больных скелетной формой и у 10 % с глазной формой Биопсия мышц - не имеет существенного диагностического значения. Находят атрофию мышечных волокон ІІ типа, признаки денервации КТ переднего средостения для выключения тимомы. Точность этого метода в диагностике тимом становит 96 %. Тимома есть у 10-15 % больных с миастенией. У большинства больных (90 %) с миастенией можно найти антитела к ацетилхолиновым рецепторам

Лечение Антихолинэстеразные препараты: калимин 30 мг трижды в день, постепенно можно дозу увеличивать до 60-120 мг (принимать во время еды). Эффект наступает через 10-30 мин, достигает максимума через 2 часа и длится еще 2 часа. Всего действие калимина может длиться до 6-8 часов. прозерин по 0,5-1,5 мг п/к (его действие кратковременно – 2-3 часа) оксазил - по 5 мг per oss. Действие длится 4-8 часов на скелетные мыщцы. Препараты К 3-4 г в сутки.

Лечение Кортикостероиды. Доза: стартовая 15-20 мг/сутки, увеличивая дозу на 5 мг каждые 2-3 дня до 50-60 мг/сут. После стабилизации состояния дозу можна уменьшать на 10 мг каждый месяц до 20-30 мг, а затем темп снижения дозы уменьшают (2,5–5 мг каждые 1-2 месяца). Поддерживающая доза 5-20 мг через день принимается в течении многих лет. Другая схема больших доз: сразу дать дозу 1-1,5 мг/кг веса для достижения эффекта (ч-з день), а затем уменьшать по ½ табл. до поддерживающей дозы. Анаболические гормоны - ретаболил 50 мг 1 раз на 3 дня 5-6 инъекций.
 назначают при генерализованной и бульбарно")
Лечение Иммуносупрессоры (азатиоприн) назначают при генерализованной и бульбарной формах от 50 мг/сутки до 150-200 мг/сутки. Клинически эффект наступает через 1,5-3 мес от начала лечения и достигает максимума до 1 года. Плазмаферез - при остром прогрессирующем течении Лучевая терапия тимуса - Показана больным преклонного возраста, перед операцией и после не полной тимэктомии. Препараты, что улучшают обменные процессы: глютаминовая кислота, метионин, вит. Е, В.
. По")
При миастеническом кризе: плазмаферез (заменяют 2-3 л плазмы 3-ды за неделю). Побочные эффекты: гипоальбуминэмия, гипотензия, нарушения гемостаза внутривенное введение иммуноглобулина (2 г/кг веса 2-5 дней) кортикостероиди (преднизолон в дозе 100 мг) в/в прозерин 1-2 мл на 20 мл 40% р-ра глюкозы ИВЛ, вдыхание кислорода, антибиотики при возбуждении галоперидол 1 мл 0,5 % р-ра в/в или в/м

Холинэргический криз Клинические признаки - фасцикуляции, судороги, брадикардия, слюнотечение, гипергидроз, боль в животе. Лечение: атропин 1 мл 0,1 % р-ра п/к или в/в дипироксим 1 мл 15 % р-ра в/м 1 раз в сутки

Противопоказаны при миастении миорелаксанты транквилизаторы антиаритмические антибиотики – аминогликозиды морфин барбитураты

Наследственные болезни с преимущественным поражением пирамидной системы: Спастическая параплегия Штрюмпеля Семейный спастический паралич с амиотрофией, олигофренией и дегенерацией сетчатки (описан Келлином) Семейный спастический паралич с ихтиозом и олигофренией (описан С’егреном и Ларссоном)

Спастическая параплегия Штрюмпеля обусловлена дегенерацией пирамидных путей, тонких пучков и мозжечковых связей. Тип наследования: частая форма – рецессивный, редкая – доминантный Клиника Начальные признаки в возрасте 10-15 лет Нижняя спастическая параплегия с резким повышением тонуса, повышением сухожильных рефлексов, патологическими рефлексами, клонусами стоп и коленных чашечек Варусная установка стоп Симметричное поражение ног Характерные признаки: Преобладание спастики над парезом Сохранение кожных рефлексов Отсутствие нарушения функции тазовых органов



Наследственные болезни с преобладающим поражением экстрапирамидной системы: Гепато-церебральная дегенерация (болезнь Вестфаля-Вильсона-Коновалова) Торзионная дистония Двойной атетоз Хорея Гентингтона Болезнь Паркинсона (дрожательный паралич) Миоклонус-эпилепсия (описана Унферрихтом и Лундборгом) Генерализированный тик Туретта Наследственное дрожание (тремор Минора) Судорога Рюльфа Пальпебро-мандибулярная синкинезия Гунна

Паркинсонизм Это хронический прогрессирующий нейродегенеративный синдром, который характеризуется двигательными нарушениями вследствие поражения экстрапирамидной системы Болезнь Паркинсона (БП) – хроническое прогрессирующее дегенеративное заболевание ЦНС, что клинически проявляется нарушением произвольных движений

ЭТИОЛОГИЧЕСКАЯ СТРУКТУРА ПАРКИНСОНИЗМА Первичный паркинсонизм болезнь Паркинсона юношеский паркинсонизм Вторичный (симптоматический паркинсонизм) сосудистый паркинсонизм медикаментозный (резерпин, антидепрессанты, бутифероны, антипсихотики, фенотиазины) токсическая энцефалопатия (отравление марганцем, ртутью, оксидом углерода) постэнцефалитический паркинсонизм метаболические энцефалопатии (печеночная недостаточность, гипотиреоз) Опухоли, гидроцефалия

ЭТИОЛОГИЧЕСКАЯ СТРУКТУРА ПАРКИНСОНИЗМА Паркинсонизм при других наследственных заболеваниях ЦНС спорадические заболевания (мультисистемная атрофия, прогрессирующий надядерный паралич, болезнь Альцгеймера, болезнь Крейцфельда-Якоба) Наследственные заболевания (болезнь Гентингтона, спино-церебелярная дегенерация, гепато-лентикулярная дегенерация)

Основные симптомы паркинсонизма І. Гипокинезия маскоподобное лицо, редкое мигание дисфагия микрография нарушения речи (монотонная, медленная) медленная походка (пропульсии) ІІ. Ригидность мышц феномен „зубчастого колеса” увеличение ригидности в момент движения поза „просителя” (ригидность в згибателях тулувища) ІІІ. Постуральные нарушения паркинсоническая „кисть” феномен „замирания” при ходьбе шаркающая походка ІУ. Тремор покоя уни- или билатеральный непроизвольный тремор, который проявляется в покое и меньше выражен во время движения движения большого и указательного пальцев по типу „счета монет”, „катания пилюль”
")
Клинические формы дрожательная ригидная смешанная Степени тяжести (по Петелину) І - незначительное снижение активности, что не отражается на трудоспособности и выполнении профессиональных обязанности ІІ – значительное снижение профессиональной и бытовой активности ІІІ – потеря возможности к самообслуживанию
 начало с изолированного аки")
Особенности дебюта начало с дрожания (у 2/3 больных) начало с изолированного акинетико-гипертонического синдрома (потеря синхронности движений рук во время ходьбы, микрография, нарушение коротких и быстрых движений рукой (чистка зубов, взбивание омлета), утомляемость при ходьбе (ощущение, будто одна нога «волочится») обманчивое начало с нарушений ходьбы (у больных с множественными очагами ишемии, при наличии пирамидных симптомов, при гидроцефалии (пациенты ходят малыми шагами с тенденцией к ретропульсии) с депрессии с боли (неврологические проявления остеохондроза, ревматизма) начало с ухудшения общего состояния, як проявление астении и утомляемости

Стратегия лечения Болезни Паркинсона защита дофаминсинтезирующих нейронов нейропротекторами и нейротрофогенами активация соответствующих процессов в дофаминергических нейронах путем улучшения их трофики активация дофаминовой трансмиссии на синаптическом уровне коррекция реаптейка (возвратного захвата) и катаболизма дофамина уменьшение дефицита дофамина (заместительная терапия) торможение патогенетического механизма – генератора патологически повышенного возбуждения в стриатум

Лечение: заместительная терапия леводопасодержащими препаратами (леводопа и ингибиторы ДДК: синемет, наком, мадопар, левокарбідопа) агонисты дофаминовых рецепторов: - эрголинового ряда (бромкриптин, роналин, парлодел) - неэрголинового ряда (проноран, мирапекс, ропинирол) селективные ингибиторы МАО-Б (селегелин, юмекс, сеган, когнитив) ингибиторы КОМТ (комтан) антагонисты Н МДА-рецепторов (амантадин, ПК-мерц неомидантан) блокаторы холиновых рецепторов (паркопан, артан, циклодол, акинетон)

Лечение: Принципы базовой терапии: Ноотропы Циннаризин Кавинтон адекватный подбор индивидуальных противопаркинсонических препаратов Хирургическое лечение: стереотаксические операции глубокая электростимуляция отдельных мозговых структур метод вибора при неэффективности консервативной терапии


 (болезнь Вестфаля-Вильсона-Коновалова) – одна")
Гепатоцеребральная дистрофяя (ГЦД) (болезнь Вестфаля-Вильсона-Коновалова) – одна из наиболее изученных наследственых форм экстрапирамидных заболеваний, связана с нарушением обмена церулоплазмина – белка плазмы крови, что содержит медь, и синтезируется в печени. Морфологически выявлено отложение меди в подкорковых ганглиях (п .Lenticularis), коре полушарий головного мозга, мозжечке, а также в печени, селезенке, радужке и хрусталике. В пораженных органах развиваются очаги размягчения и склерозирования Тип наследования: аутосомно-рецессивный, встречается однаково часто у мужчин и женщин

Клиника первые проявления болезни проявляются в детском или юношеском возрасте постепенное нарастание ригидности мыщц разнообразные гиперкинезы (хореиформные, торзионно-дистонические, атетоидные) изменения психики могут наблюдаться судорожные приступы увеличение печени кольцо Кайзера-Флейшера (золотисто-зеленого или зеленовато-коричневого цвета) на радужке

Неврологические формы заболевания: По классификации Коновалова Н.В. ригидно-аритмокинетическая дрожательно-ригидная дрожательная экстрапирамидно-корковая Диагноз. Опорными пунктами для постановки диагноза являются: тщательное изучение родословной наличие типичных симптомов: кольцо Кайзера-Флейшера поражение печени пониженное содержание церулоплазмина в крови повышенное содержание меди в моче (гиперкупрурия)
 Патоморфологически в основ")
Торсионная дистония (деформирующая мышечная дистония) Патоморфологически в основе дегенеративные изменения в подкорковых ганглиях, субталамических ядрах и зубчатом ядре мозжечка, нарушение синтеза и обмена медиаторов в базальных ганглиях Тип наследования: гиперкинетическая форма торсионной дистонии аутосомно-доминантный тип (хромосома 9) ригидная форма – аутосомно-рецессивный тип (хромосома 14)

Клиника начало в молодом возрасте постоянно прогрессирует гиперкинезы, которые возникают при любой попытке движения или изменения положения туловища гиперкинезы мышц туловища и мышц конечностей, которые принимают неприродные позы гиперкинез мышц шеи - спастическая кривошея, что очень часто есть первым симптомом болезни интеллект как правило не страдает локальные гиперкинезы – спастическая кривошея и пишущий спазм Диагностика. Анализ родословной больного и оценка динамики патологического процесса.

Спастическая кривошея


Хорея Гентингтона одно из тяжелейших и постоянно прогрессирующих наследственных заболеваний нервной системы, обусловленной системной дегенерацией экстрапирамидных двигательных структур и коры головного мозга. Тип наследования: аутосомно-доминантный с высокой пенетрантностью мутантного гена (хромосома 4)

Клиника начало в зрелом возрасте и очень редко у детей одинаково часто болеют мужчины и женщины Симптомы классической формы: хореиформные гиперкинезы экстрапирамидная ригидность постоянно нарастающая деменция Редкие варианты: акинетико-ригидный синдром экстрапирамидная обездвиженность у детей эпилептиформные приступы миоклонии


Наследственные болезни с преобладанием поражения мозжечковой системы (наследственные атаксии): Спинальная атаксия Фридрайха наследственная мозжечковая атаксия Пьєра Мари Разные клинические формы оливо-понто-церебелярной дегенерации Болезнь Рефсума Болезнь Руси-Левина Болезнь Маринеску-Шагрена Болезнь Лихтенстейна- Кнорра

Спинальная атаксия Фридрейха дегенерация спинного мозга, дегенеративно-дистрофические изменения в задних и боковых канатиках Тип наследования: аутосомно-рецесивный (хромосома 9)

Клиника начало в 10-12 летнем возрасте медленно прогрессирует сенситивно-мозжечковая атаксия (нистагм, табетично-мозжечковая походка, скандированная речь, атаксия туловища и верхних конечностей гипотония мышц и арефлексия вначале заболевания выпадает вибрационное и глибокое мышечно-суставное чувство на нижних конечностях по проводниковому типу аномалии развития костей „дизрафический статус”: кифосколиоз, стопа Фридрейха признаки кардиомиопатии характерно снижение интеллекта на поздних стадиях заболевания присоединяются признаки поражения пирамидных путей

Атаксия Фридрейха Pes cavus

Наследственная мозжечковая атаксия Пьєра Мари начало болезни на 3-4 десятилетии жизни постоянно прогрессирующее течение мозжечковая атаксия дизартрия гиперрефлексия, спастическая гипертония мышц в ногах, патологические рефлексы атрофия зрительных нервов, дегенерация сетчатки и глазодвигательные нарушения изменения психики (снижение интеллекта, памяти и нарушения в эмоционально-волевой сфере) Тип наследования: аутосомно-доминантный

Оливопонтоцеребелярная дегенерация группа заболеваний, которые характеризуются большой клинической и генетической гетерогенностью. они связаны с системным поражением нейронов: коры мозжечка ядер моста мозга нижних олив клеток передних рогов спинного мозга базальных ганглиев Тип наследования: аутосомно-доминантный с неполным проявлением, есть указания на рецесивный тип наследования

Наследственные болезни с преимущественным поражением нервно-мышечной системы: І. Прогрессирующие мышечные дистрофии: Детская псевдогипертрофическая мышечная дистрофия Дюшенна Позняя псевдо-гипертрофическая мышечная дистрофия Беккера Миодистрофия Эмери- Дрейфуса Семейная висцеральная миопатия Плече-лопатково-лицевая форма Ландузи-Дежерина Лопатково-перонеальна форма Давиденкова Ювенильная мышечная дистрофия Эрба Офтальмоплегическая миопатия Дистальная миопатия Валендера Врожденные непрогрессирующие формы миопатии




ІІ. Амиотрофии как результат поражения периферического двигательного нейрона: Детская спинальная амиотрофия Верднига-Гоффмана Юношеская проксимальная амиотрофия Кугельберга-Веландера Поздняя спинальная и бульбарная амиотрофия Кеннеди Невральная амиотрофия Шарко-Мари-Тутта Гипертрофический неврит Дежерина-Сотта





Наследственно семейные миотонии: Миотония Томсена Атоничная миотония (миотоническая дистрофия) Парамиотония Эйленбурга Синдром Шварта-Джампела (хондродистрофическая миотония)



Наследственные нерно-мышечные заболевания с пароксизмальными состояниями: Пароксизмальная семейная миоплегия Эпизодическая наследственная адинамия Гамсторп Болезнь Мак-Ардла Эпизодическая миотоническая адинамия Беккера











