











, бледный шар и черную субстанцию. Базальные ганглии получают импульсы от лобной коры, ответственной за контроль произвольных движений, и опо…")

 не воспроизводятся. Однако ни в одном из проведенных исследований не было достоверно показано, чт…")


. Полная реакция: L-ДОФА + пиридоксальфосфат → дофамин + пиридоксальфосфат + CO2 Таким образом, для синтеза дофамина необходим также п…")





. Существу…")


 нейронов мозга, две независимых лаборатории получили интересные результаты о причинах старения и болезни Паркинсона Объектом исследований был голо…")
 старых инди…")


 –белка, имеющего отношение к репарации клеточных ме…")


 типа В; - агонисты дофаминовых рецепторов; - ингибиторы транспорта дофамина; - антагонисты возбуждаю…")
 провела комплексное исследование микроРНК м…")

 Болезнь Хантингтона является одним из видов нейродегенеративных заболеваний, и поражает примерно одного из каждых ста тысяч человек, согласно статистике по Западным странам. Заболевание носит наследственный характер…")





Презентация на тему: STARIUS

Лекция 7. Болезнь Паркинсона

Определение Болезнь Паркинсона - хроническое прогрессирующее дегенеративное заболевание центральной нервной системы, клинически проявляющееся нарушением произвольных движений. Впервые описано в 1817 году Джеймсом Паркинсоном в книге "Эссе о дрожательном параличе". Болезнь Паркинсона или паркинсонизм – заболевание пожилых людей, чаще всего развивается в возрасте 70-80 лет. К сожалению, в последние десятилетия болезнь Паркинсона неуклонно «молодеет», и поэтому нередки случаи возникновения паркинсонизма на шестом десятилетии жизни и даже раньше.

Характерные симптомы Для болезни Паркинсона характерны следующие симптомы : дрожание "скованность" мышц замедленность движений неустойчивое равновесие. Кроме того, у больных с болезнью Паркинсона утрачивается выразительность мимики. Они могут выглядеть равнодушными, безразличными, хотя на самом деле переживают ту или иную ситуацию как здоровые люди. Сходные симптомы наблюдаются также при поражениях головного мозга в случае гипертонической болезни и/или атеросклероза, после травмы (повторных травм) головного мозга и некоторых других хронических заболеваниях головного мозга.

Другие характерные симптомы Вовлечение речедвигательной мускулатуры приводит к нарушению речи - она может стать мало модулированной, неразборчивой Нарушение движений изменяет и почерк. Если больной не старается хорошо писать, то нередко бывает трудно прочитать написанное. Кроме перечисленных нарушений движения бывают и так называемые вегетативные расстройства: изменение аппетита, слюноотделения, деятельности кишечника (запоры), частое мочеиспускание При болезни Паркинсона у больных часто наступает депрессия. Это связано с двумя причинами: во-первых, больной бывает огорчен тем, что не может действовать так же полноценно, как прежде ни на работе, ни дома; во-вторых, при этом заболевании происходит повреждение тех отделов мозга, которые обеспечивают человеку уравновешенное настроение. Проявления депрессии разнообразны, но включают в себя ряд характерных симптомов: плохое настроение, необычно быстрая утомляемость, снижение внимания и сосредоточенности, тревога и раздражительность, безразличие к окружающему, снижение аппетита.

Течение болезни Паркинсона Средний возраст начала болезни Паркинсона - 55 лет. В то же время у 10% больных заболевание начинается в относительно молодом возрасте, до 40 лет. Заболеваемость паркинсонизмом не зависит от половой и расовой принадлежности, социального положения и места проживания. Предполагают, что с увеличением среднего возраста населения в ближайшие годы распространенность болезни Паркинсона в популяции будет возрастать. На поздних стадиях заболевания качество жизни больных оказывается существенно сниженным. При грубых нарушениях глотания пациенты быстро теряют в весе. В случаях длительной обездвиженности смерть больных обусловлена присоединяющимися дыхательными расстройствами и пролежнями.

Распространенность В России насчитывается до 350000 больных болезнью Паркинсона. В США болезнью Паркинсона страдает около 500000 человек. После деменции, эпилепсии и сосудистых заболеваний мозга болезнь Паркинсона является наиболее частой проблемой пожилых людей, о чем свидетельствует ее распространенность в России (данные 1996 года): 1,8 : 1000 в общей популяции 1,0 : 100 в популяции тех, кому за 70 1,0 : 50 в популяции тех, кому за 80

Этиология Происхождение болезни Паркинсона остается до конца не изученным, тем не менее в качестве причины заболевания рассматривается сочетание нескольких факторов: старение наследственность некоторые токсины и вещества.

Причины болезни Паркинсона Старение Тот факт, что некоторые проявления болезни Паркинсона возникают и при нормальном старении, заставляет предполагать, что одной из причин паркинсонизма может быть возрастное снижение активности нейронов мозга. Наследственность Уже в течение многих лет обсуждается возможность генетической предрасположенности к болезни Паркинсона и накоплено много информации о наличии мутантных генов, вовлеченных в развитие болезни Токсины и другие вещества В 1977 году было описано несколько случаев тяжелого паркинсонизма у молодых наркоманов, принимавших синтетический героин. Этот факт свидетельствует о том, что различные химические вещества могут "запускать" патологический процесс в нейронах головного мозга и вызывать проявления паркинсонизма. Известен также марганцевый паркинсонизм, ставший в последние годы серьезной проблемой в связи с употреблением лицами преимущественно молодого возраста суррогатных наркотических соединений, содержащих марганец.

Другие причины Причины болезни Паркинсона включают также: вирусные инфекции, приводящие к постэнцефалитному паркинсонизму; атеросклероз сосудов головного мозга; тяжелые и повторные черепно-мозговые травмы. Длительный прием некоторых препаратов, блокирующих освобождение или передачу дофамина (например нейролептиков, средств, содержащих резерпин), может также привести к появлению симптомов паркинсонизма, поэтому всегда следует уточнять, какую терапию получал больной до установления диагноза болезнь Паркинсона.

Течение заболевания Как правило, болезнь Паркинсона имеет медленное течение, так что на ранних стадиях заболевание может не диагностироваться в течение ряда лет

Регуляция движения и болезнь Паркинсона В осознанном контроле за произвольными движениями принимает участие кора лобных долей головного мозга, откуда нервные импульсы передаются в конечности. Одним из наиболее важных нейротрансмиттеров в этой области является ацетилхолин. В акте произвольного движения участвует и так называемая пирамидная система, представленная эфферентными нейронами, тела которых располагаются в коре большого мозга, оканчиваются в двигательных ядрах черепных нервов. Однако важным для целостного процесса движения является контроль не только за произвольными, но и за непроизвольными движениями. Эту функцию выполняет экстрапирамидная система, обеспечивающая плавность движений и возможность прервать начатое действие. Эта система объединяет структуры - базальные ганглии, располагающиеся за пределами продолговатого мозга (отсюда термин "экстрапирамидная система”). Нейротрансмиттером, обеспечивающий бессознательный контроль за движениями, является дофамин. В основе ,олезнь Паркинсона лежит снижение активности дофаминергических нейронов черной субстанции.

Базальные ганглии Базальные ганглии объединяют структуры: хвостатое ядро, скорлупу (вместе - полосатое тело), бледный шар и черную субстанцию. Базальные ганглии получают импульсы от лобной коры, ответственной за контроль произвольных движений, и опосредуют обратный непроизвольный контроль за движениями через премоторную кору и таламус.

Регуляция мышечного сокращения и медиаторы Первичные симптомы болезни Паркинсона обусловлены чрезмерной сократимостью мышц Мышечные сокращения контролируются ацетилхолином через 5 типов холинергических рецепторов: m1, m2, m3, m4 и m5. Рецепторы m1, m3 и m5 – стимулирующие, m2 и m4 - ингибирующие. Комбинированный эффект m1, m3 и m5 является более сильным, чем эффект m2 и m4. Таким образом, в целом ацетилхолин стимулирует сокращение. Дофамин влияет на сокращение, также воздействуя на 5 типов рецепторов: D1, D2, D3, D4 и D5. Рецепторы D2, D3 и D4 – ингибирующие, D1 и D5 -стимулирующие. Комбинированный ингибирующий эффект D2, D3 и D4 является более сильным, чем комбинированный стимулирующий эффект от D1 и D5. Суммарный эффект дофамина –это ингибирование мышечного сокращения. Болезнь Паркинсона связана с тем, что эффект дофамина выражен меньше, чем эффект ацетилхолина. Это вызвано дефицитом дофамина в черной субстанции.

Цитологические данные Дофамин производится в дофаминергических нейронах, которых в мозге около 7 тысяч. Дофаминергические нейроны (как и многие другие) не воспроизводятся. Однако ни в одном из проведенных исследований не было достоверно показано, что болезнь Паркинсона связана с потерей именно дофаминергических нейронов, зато было продемонстрировано существенное снижение активности дофаминергических нейронов, что часто вызвано снижением активности ферментов, обеспечивающих синтез дофамина.

Синтез дофамина и адреналина

Синтез дофамина Первопричиной болезни Паркинсона является дефицит дофамина. Дофамин синтезируется в дофаминергических нейронах благодаря следующим реакциям: L-тирозин → L-ДОФА → дофамин Первая стадия биосинтеза осуществляет тирозингидроксилаза, которая лимитирует скорость всего процесса. L-тирозин + ТГФ + O2 + Fe2+ → L-ДОФА + DHFA + H2O + Fe2+ Таким образом, для образования L-ДОФА необходимы, L-тирозин, TГФ (тетрагидрофолевая кислота) и ионы железа. Активность тирозингидроксилазы при болезни Паркинсона падает до 25% от нормы, а в тяжелых случаях – до менее чем 10%.

Синтез дофамина На второй стадии синтеза участвует декарбоксилаза ароматических L-аминокислот (ДОФА декарбоксилаза). Полная реакция: L-ДОФА + пиридоксальфосфат → дофамин + пиридоксальфосфат + CO2 Таким образом, для синтеза дофамина необходим также пиридоксальфосфат. Активность ДОФА-декарбоксилазы при болезни Паркинсона может составлять менее 25% от нормального уровня.

Коферменты, необходимы для синтеза дофамина Для синтеза дофамина необходимо наличие трех коферментов: тетрагидрофолата, пиридоксальфосфата и НАДН, который необходим для образования TГФ и пиридоксальфосфата из предшественников : Фолиевая кислота → дигидрофолиевая кислота → тетрагидрофолиевая кислота Пиридоксин → пиридоксаль → пиридоксаль 5-фосфат (здесь в качестве кофактора нужен цинк)

Дефекты в синтезе дофамина Первичным событием, обеспечивающим деградацию клеток при болезни Паркинсона, может быть отсутствие синтеза L-ДОФА L-тирозин + THFA + O2 + Fe2+ → L-ДОФА + DHFA + H2O + Fe2+ Если отсутствует TГФ, то происходит следующее : L-тирозин + Fe2+ + O2 → L-тирозин + Fe3+ + O-2 (супреоксиданион) Супероксианион – один из наиболее деструктивных элементов в клетке. Образование ДОФА не происходит, если отсутствует L-тирозин. Таким образом, недостаток компонентов, участвующих в синтезе дофамина (L-тирозин, TГФ и ионы железа), могут быть причиной снижения активности дофаминергических нейронов)

Антиоксиданты Витамин C и витамин E были использованы для предотвращения деградации активности дофаминергических рецепторов клеток при болезни Паркинсона. Разрушение супероксида осуществляется ферментом супероксиддисмутазой: 2O2 + 2H+ → H2O2 + O2 Образующая перекись разрушается каталазой H2O2 → H2O + 1/2 O2 Антиоксиданты витамин C и витамин E могут замедлить дегенерацию дофаминэергических нейронов, но они не устраняют основной проблемы, в результате которой образуется супероксид-анион.

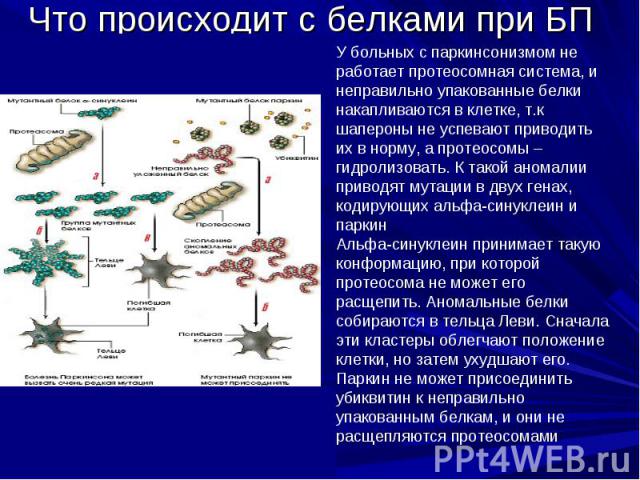
Тельца Леви При болезни Паркинсона в цитоплазме дофаминергических нейронов образуются тельца Леви, которые описываются как агрегаты, содержащие остатки клеток. Они являются симптомом, свидетельствующим о распаде клеток. Состоят из плотно агрегированных филаментов включающих убиквитин, альфа-синуклеин и белки нейрофиламентов. Однако тельца Леви не уникальны для болезни Паркинсона, они встечаются и при других нейродегенеративных заболевания, в частности, при болезни Альцгеймера.

Мутации в белке-паркине Показано, что белок паркин является важнейшим звеном системы клеточной защиты и, в частности, непосредственно участвует в деградации α-синуклеина – классического белкового маркера болезни Паркинсона в составе характерных интранейрональных включений (телец Леви)


Связь между старением и болезнью Паркинсона Старение — результат действия множества вредных генетических признаков, которые проявляться лишь по окончании репродуктивного периода, DILL-признаки (DILL — от Deleterious In Late Life). Существует несколько причин накопления DILL-признаков в течение эволюции. Во-первых, численность популяции на протяжении большей части эволюции человека была низкой (около 10 000), поэтому случайные процессы сильно влияли на эволюцию генов и приводили к закреплению многих вредных аллелей. Недавнее резкое увеличение численности популяции еще не отразилось на эволюции генов человека. Во-вторых, мутация, проявляющаяся лишь после репродуктивного периода, не влияет на число детей и поэтому может распространиться в популяции случайным образом без влияния естественного отбора. В-третьих, даже если бы DILL-мутации влияли на приспособленность, естественный отбор не мог элиминировать эти мутации, поскольку средняя продолжительность жизни была низкой. Все они не влияли на развитие популяции, пока примерно 10 000 лет назад человек не занялся земледелием, в результате чего увеличилась продолжительность жизни. Тогда DILL-мутации стали проявляться. Начиная с этого момента человек стал умирать преимущественно от внутренних причин, а не от внешних.

Связь между старением и болезнью Паркинсона Гены накапливали DILL-мутации неравномерно, поэтому и потенциальная вредность этих генов в старости различна. Митохондриальные гены, наследуемые через цитоплазму яйцеклетки от матери, должны содержать много DILL-мутаций. В связи с гаплоидностью, цитоплазматическим наследованием и отсутствием рекомбинации митохондриальные гены обладают низкой численностью, поэтому на их эволюцию сильно влияют случайные факторы и слабо влияет естественный отбор. В результате дрейфа генов в малочисленной популяции митохондриальных генов могут фиксироваться аллели с вредными мутациями. Митохондрии обладают повышенным темпом мутирования, что увеличивает вероятность появления новой соматической мутации в течение жизни человека. Но, поскольку в каждой клетке содержатся тысячи митохондриальных геномов (около 2000), то один появившийся мутант, сосуществуя с нормальными митохондриальными геномами, может не сильно влиять на приспособленность клетки. Дальнейшая судьба мутантного генома зависит от соотношения сил на внутриклеточном и межклеточном уровнях

Связь между старением и болезнью Паркинсона Внутри клетки митохондриальные геномы конкурируют друг с другом. Геномы, которые быстрее реплицируются, вытесняют медленно-размножающихся конкурентов. Если у одного генома произошла крупная делеция, то он становится нефункциональным (так как это приводит к производству меньшего количества АТФ), но зато коротким и быстро размножающимся. У такого генома хорошие шансы на победу во внутриклеточной борьбе. Однако клетка, в которой мутантные митохондриальные геномы достигли высокой концентрации, менее жизнеспособна и медленнее делится по сравнению с клеткой с нормальными митохондриальными геномами, и мутантные геномы элиминируются из организма вместе несущей их клеткой. Ткани, сильно зависящие от митохондриального метаболизма (мышцы, сердце и нервная система), более чувствительны к накоплению мутантных геномов. Помимо высокой потребности в энергии нервная ткань характеризуется медленным делением клеток, что приводит к уменьшению уровня отбора митохондриальных геномов, вследствие чего может проявиться патогенный эффект, вызванный мутантными митохондриальными геномами.

Связь между старением и болезнью Паркинсона Исследуя мутации в митохондриальной ДНК (мтДНК) нейронов мозга, две независимых лаборатории получили интересные результаты о причинах старения и болезни Паркинсона Объектом исследований был головной мозг умерших людей, а именно substantia nigra. Исследования митохондрий в нейронах черной субстанции показали быстрое накопление делеций с возрастом. Оказалось также, что некоторые нейроны имеют сильный дефицит цитохром c оксидазы (COX). Анализ показал, что нейроны черной субстанции у людей, погибших от болезни Паркинсона, содержат более 60% мутантной мтДНК, и делеции находятся в области генов, кодирующих субъединицы цитохром c оксидазы.

Связь между старением и болезнью Паркинсона Разные нейроны содержали уникальные делеции, что говорит о соматическом происхождении этих мутаций в нейронах. Интересно, что нейроны, выделенные из соседней области головного мозга (гиппокамп) старых индивидуумов не показывали высокого уровня делеций. Возможно, активность нейронов черной субстанции сопряжена с образованием активных форм кислорода, которые, являясь мутагенами, приводят к образованию делеций в мтДНК. Активные формы кислорода могут появляться также и при метаболизме дофамина. Основываясь на этом предположении, для предотвращения появления делеций врачи предлагают использовать антиоксиданты. Итак, соматические делеции в мтДНК играют важную роль в деградации нейронов при старении и болезни Паркинсона. Поскольку делеции появляются независимо в разных нейронах черного вещества примерно в одно и тоже время и затрагивают примерно одно и то же место, скорее всего существуют определенные DILL-мутации, индуцирующие эти делеции. Например, в мтДНК человека существует повтор из 13 пар оснований, который приводит к делециям в соматических тканях, причем индуцирует именно делеции, затрагивающие гены цитохром-c-оксидазы. Этот повтор (как и все повторы в мтДНК) можно интерпретировать как DILL-мутацию

Связь между старением и болезнью Паркинсона Другая потенциальная DILL-мутация может находиться в митохондрильной ДНК-полимеразе — ферменте, обеспечивающем репликацию мтДНК. Мутантная форма данного фермента приводит к большому числу ошибок при репликации мтДНК, и линии мышей с таким ферментом страдают от раннего старения из-за накопления большого числа мутаций в мтДНК. Скорее всего, болезнь Паркинсона, как и старение, может быть вызвана многочисленными причинами. Например, мутация в кодируемом в ядре гене альфа-синуклеина приводит к слипанию мутантных белков и образованию цитоплазматических включений, характерных для болезни Паркинсона. Сходный эффект проявляется при нарушении у мышей автофагии — процесса удаления старых белков из цитоплазмы клеток.

Уровень холестерина и болезнь Паркинсона Люди, имеющие низкий уровень холестерина ЛПНП в крови, чаще заболевают болезнью Паркинсона. Анализ содержания холестерина в крови у 236 человек, 124 из которых страдали болезнью Паркинсона, показал, что у людей, имеющих низкий уровень холестерина ЛПНП болезнь Паркинсона развивалась более чем в 3,5 раза чаще, чем у лиц с высоким уровнем ЛПНП. Поскольку уровень холестерина в значительной степени обусловлен наследственностью, это может свидетельствовать об одном из факторов, посредством которого наследственность может влиять на развитие болезни Паркинсона. Однако почему низкий уровень холестерина способствует развитию болезни –остается неясным

Мутации в белках, приводящие к БП Известным модификатором возраста начала болезни Паркинсона и ряда других нейродегенеративных заболеваний является генетический полиморфизм аполипопротеина Е (ароЕ) –белка, имеющего отношение к репарации клеточных мембран и мобилизации “строительного” холестерина в ряде работ было установлено, что наличие аллеля ароЕε4 способствует более раннему началу болезни Паркинсона [2, 47]. В наибольшей степени этот эффектпроявляется при гомозиготности по данному варианту ароЕ (генотип ε4/ε4): в таких случаях возраст начала болезни составил 35–37 лет.

Гены, дефекты в которых приводят к БП Показана ассоциация болезни Паркинсона с рядом полиморфизмов в генах детоксикации ксенобиотиков, системы антиоксидантной защиты клетки, транспорта и метаболизма дофамина, липидного обмена, митохондриального цикла. Гены систем клеточной детоксикации и антиоксидантной защиты • параоксоназа1 • убиквитин-С-концевая гидролаза L1 • цитохром Р450 (CYP2D6) • N-ацетилтрансфераза 2 • семейство ферментов глутатинтрансферазы • гемоксигеназа1 • ферменты α-кетоглутаратдегидрогеназного комплекса • супероксиддисмутаза

Гены, дефекты в которых приводят к БП Гены транспорта и метаболизма дофамина • моноаминоксидазы А и B • катехол-О-метилтрансфераза • тирозингидроксилаза • транспортеры дофамина • дофаминовые рецепторы D2, D3, D4 и D5 Митохондриальный геном • тРНКГлу • митохондриальная ДНК (отдельные полиморфизмы) • комплекс I электронной дыхательной цепи • цитохром с оксидаза Другие гены • NO-синтазы (nNOS, iNOS) • аполипопротеин Е • нейротрофические факторы Носительство неблагоприятных аллельных вариантов данных генов достоверно повышает риск заболевания т.е. формирует генетическую предрасположенность к болезни Паркинсона,

Лечение болезни Паркинсона Средства с антиоксидантным эффектом (альфа-токоферол, тиоктовая кислота, десфероксамин, ингибиторы моноаминоксидазы (МАО) типа В; - агонисты дофаминовых рецепторов; - ингибиторы транспорта дофамина; - антагонисты возбуждающих аминокислот (амантадин, будипин, ремацемид, ри-лузол); - трофические факторы (глиальный нейротрофический фактор, мозговой фактор роста, фибробластный фактор роста); - противовоспалительные средства (ингибиторы синтетазы азота, иммунофилины, талидомид).

Возможная роль микроРНК в развитии болезни Паркинсона Команда американских ученых из Колумбийского университета в Нью-Йорке и Уотсоновской биологической школы Лаборатории Колд-Спринг-Харбор (штат Нью-Йорк) провела комплексное исследование микроРНК мозга у больных паркинсонизмом. Они выявили 224 различных микроРНК, свойственных лишь нервным клеткам. Из этого набора концентрация одной микроРНК, а именно miR-133b, у больных паркинсонизмом оказалась значительно меньше, чем у здоровых людей. Ученые нашли, что miR-133b специфически связывается с транскрипционным фактором Pitx3, от которого зависит созревание дофаминэргических нейронов. Если в клетках мышей нет этого белка, то и нейроны черной субстанции не развиваются, как это было показано несколькими годами раньше другой группой ученых того же Колумбийского университета. Согласно предположению Азы Абелиовича (Asa Abeliovich; см. также любопытную статью о нём: The California Stem-Cell Gold Rush) с коллегами, существует обратная регуляторная петля miR-133b и Pitx3: если микроРНК много, она связывается с транскрипционным фактором Pitx3, он перестает работать, и транскрипция (то есть синтез) miR-133b останавливается. В результате концентрация miR-133b снижается; тогда высвобождается Pitx3 и активирует выработку miR-133b, и концентрация этих молекул повышается. Таким образом, регуляторная петля замыкается. Это означает, что в нормально работающей клетке концентрация обеих молекул — и miR-133b, и Pitx3 — должна поддерживаться на каком-то постоянном, строго определенном уровне.

Роль диеты в профилактике нейродегенеративных заболеваний Согласно результатам недавнего исследования, регулярное употребление рыбы, фруктов и овощей предотвращает развитие нейродегенеративных заболеваний, в частности, деменции. При этом риск снижается на 60%, отмечают специалисты. Ученые утверждают, что рацион питания, богатый рыбой, жирными кислотами ряда омега-3, фруктами и овощами, гарантирует защиту человека от развития деменции и болезни Альцгеймера, тогда как частое потребление омега-6 жирных кислот угрожает ухудшением памяти в зрелом возрасте. Об этом заявляют ученые из Национального университета медицинских исследований Франции
 Болезнь Хантингтона является одним из видов не")
Болезнь Гентингтона (Хантингтона) Болезнь Хантингтона является одним из видов нейродегенеративных заболеваний, и поражает примерно одного из каждых ста тысяч человек, согласно статистике по Западным странам. Заболевание носит наследственный характер: если один из родителей страдает болезнью Хантингтона, то шансы новорожденного ребенка оказаться ее жертвой составляют 50 процентов

Болезнь Гентингтона Болезнь Гентингтона - аутосомно-доминантное заболевание, проявляющееся деменцией и двигательными нарушениями. В тех случаях, когда болезнь начинается в зрелом возрасте, она характеризуется симптомами гиперактивности дофаминергической системы. Главный из этих симптомов - хорея (быстрые, отрывистые движения, обычно конечностей, иногда напоминающие произвольные). Ей нередко сопутствует снижение мышечного тонуса. При болезни Гентингтона и других экстрапирамидных заболеваниях хорея часто сочетается с атетозом - медленными и плавными червеобразными движениями, которые меньше напоминают произвольные. Иногда рассматривают как единый гиперкинез (хореоатетоз). В детском возрасте болезнь Гентингтона может проявляться паркинсонизмом. К основным проявлениям относят также прогрессирующие эмоциональные расстройства, изменения личности и деменцию. Нередко наблюдается депрессия; примерно 5% больных заканчивают жизнь самоубийством Обнаружен лежащий в основе заболевания генетический дефект (повторяющаяся последовательность нуклеотидов в 4-й хромосоме). Сходный гиперкинез может возникать при других заболеваниях, поражающих базальные ядра: отравлении ртутью, ревматизме (хорея Сиденгама), инфекциях (дифтерии, коклюше, краснухе, прочих вирусных энцефалитах и др.),приеме противосудорожных средств, лития, противорвотных средств, при беременности (редко), тиреотоксикозе, билирубиновой энцефалопатии, сенильной хорее и при других заболеваниях. Лечение. На ранней стадии используют средства, истощающие запасы дофамина или блокирующие дофаминовые рецепторы.

Рассеянный склероз и боковой амиотрофический склероз Рассеянный склероз - хроническое прогрессирующее заболевание нервной системы, особенностью которого является одновременное поражение нескольких различных отделов нервной системы, что приводит к появлению у больных разнообразной неврологической симптоматики, часто приводящее к инвалидизации больного. Рассеянный склероз может прогрессировать десятилетиями, приводя постепенно к все большей инвалидности больного, а боковой амиотрофический склероз обычно заканчивается летальным исходом за несколько лет или даже месяцев

Возможное участие фибрина в развитии рассеянного склероза Белок фибрин, вовлеченный в свёртывание крови, также играет важную роль в подстрекательном ответе и развитии ревматического артрита, сообщают учёные университета Цинциннати /США/. Согласно их исследованию, болезнь вызвана взаимодействием подстрекательных клеток с фибрином через рецептор aMB2. Врачи полагают, что фармакологическое запрещение связи белка и рецептора может эффективно предотвратить формирование артрита и рассеянного склероза. Ревматический артрит - болезненная и изнурительная болезнь, вовлекающая хроническое воспаление, вырождение костной ткани, потерю хряща и в конечном счете трудность самостоятельного передвижения. Хотя точная причина болезни полностью не известна, медики предполагают, что активация определенных компонентов в иммунной системе определяет формирование и раннюю прогрессию артрита. Накопления фибрина - отличительная особенность поврежденных суставов, и, как говорят специалисты, белок связан с процессом их воспаления.

Роль вирусов и генетической предрасположенности в развитии рассеянного склероза Пусковым механизмом рассеянного склероза, в основе которого лежит необратимое разрушение миелина (вещества, образующего защитный слой вокруг нервных волокон и проводящего нервные импульсы), является медленно текущая вирусная инфекция, вызывающая иммунную агрессию, направленную на собственный организм человека, на его нервную систему. На подозрении у специалистов более десятка вирусов — кори, краснухи, простого герпеса, разные виды ретровирусов. Однако основной виновник болезни пока не выявлен. Согласно другой версии, рассеянный склероз — генетически обусловленное заболевание. Основанием для такого предположения стал тот факт, что загадочный недуг в два раза чаще встречается у людей белой расы, живущих в северных широтах, чем у южан и представителей других рас. Но единого гена, определяющего предрасположенность к рассеянному склерозу, ученые тоже не нашли.

Последствия рассеянного склероза если рассеянный склероз вовремя не затормозить, печальный исход неизбежен. Заболевание развивается медленно, но неумолимо. Через 10–15 лет болезни более 50% больных уже не могут ходить, сидеть. Затем следуют слепота и полная неподвижность.











