

 — прогрессирующее нейродегенеративное заболевание, характеризующееся прояв…")





, являются")
, так и более поздний (21-40 лет). До появления первых признаков заболевания большинство больных развивается нормаль…")

. …")
;инсультоподибные эпизоды;судороги;")
 или магнитно - резонансной томографии (МРТ) головного мозга;атаксия;коматозные состояния;атрофия зрительных нервов;пигментный ретинит;синдром …")


. В обоих полушариях в области лентикулярных ядер и зрительных бугров определяются очаги повышен…")
 в височной области правого полушария (а) и высокой перфузии (красный цвет) в затылочных долях (б) и височно…")
 обнаруживается мутация в нуклеотиде 3243, где происходит замена аденина на гуанин в гене транспортной РНК для лейцина. Мут…")
, витамины К1, К3 - филлохинон (25 мг / сутки) и менадион (до 75 мг / сут), янтарная кислота (до 6 мг / сут), витамин С (2-4 г / сут) и другие …")
, который обладает свойством усиливать энергетический метаболизм в мозговой ткани. Введение витаминов рибофлавина (100 мг / сутки) и никотинамида (до 1 г / …")

), сахарный диабет, гипопаратиреоз, сердечные блокады, антифосфолипидный синдром, дефицит карнитина. Значи…")

Презентация на тему: Синдром MELAS

ГУ «Крымский государственный медицинский университет им.С.И. Георгиевского»Кафедра педиатрии с курсом детских инфекционных болезней Синдром MELAS Выполнила студентка 503 группы I Медицинского факультета Могилевская А.А.

Синдром MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды») — прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями.

История Синдром MELAS впервые выделен в нозологически самостоятельную форму S. Pavlakis и соавт. в 1984 г.. Однако есть основания предполагать, что заболевание было описано раньше под названием "семейная полиодистрофия, митохондриальная миопатия, лактацидемия". К 1994 г. в литературе опубликовано 110 наблюдений синдрома MELAS.

Распространенность В зависимости от популяции частота колеблется от 10,2 на 100000 до 16,3 на 100000 взрослого населения.

Этиология, патогенез MELAS относится к митохондриальным заболеваниям. Синдром розвивается в результате точечных мутаций митохондриальной ДНК. Выявлена локализация 3 точечных мутаций, с которыми ассоциирован синдром MELAS: две - в транспортной РНК и одна - в цитохром с- оксидазе.

Соотношение полов

Тип наследования Чаще синдром наследуется по материнской линии, за исключением новых мутаций, которые впервые возникли в семье.

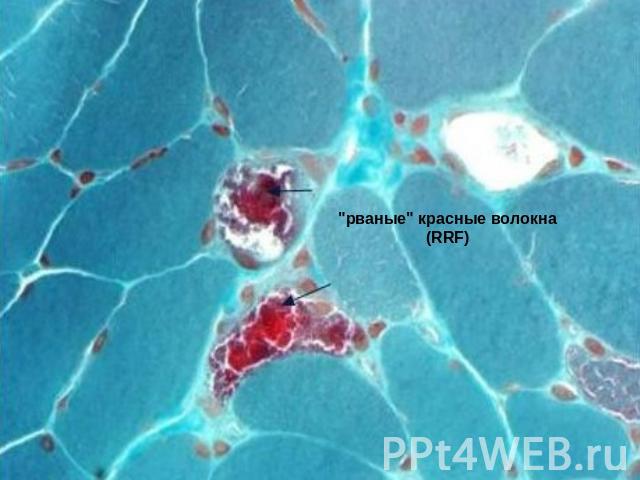
Патоморфологические изменения Характерным патоморфологическим признаком синдрома MELAS, как и ряда других митохондриальных энцефаломиопатий (синдромов Кернса - Сейр, MERRF и др.), являются "рваные" красные волокна (RRF), которые проявляются в мышечной ткани при модифицированном окраске трихромом по Гомори. Они являются морфологическим субстратом повреждения митохондриальной ДНК и образуются вследствие пролиферации аномальных митохондрий. Красные "рваные" волокна являются следствием мутаций, повреждающих гены транспортной РНК и приводят к нарушению внутримитохондриального синтеза белка. Показано, что такие морфологические характеристики мышечной ткани, как наличие сосудов с высокой активностью сукцинатдегидрогеназы и значительное количество цитохром с-оксидазопозитивних мышечных волокон, является характерной особенностью синдрома MELAS, которые позволяют дифференцировать его от синдромов Кернса-Сейр и MERRF. Одной из наспецифичниших признаков повреждения мозга при данной болезни является наличие старых и новых очагов инфарктов.
")
"рваные" красные волокна (RRF)

Клиническая картина Первые признаки чаще появляются в возрасте 6-10 лет, хотя возможны как более раннее начало заболевания (до 2 лет), так и более поздний (21-40 лет). До появления первых признаков заболевания большинство больных развивается нормально. Начальные клинические проявления: судороги, рецидивирующие головные боли, рвота, анорексия, непереносимость физической нагрузки, психические нарушения, неврологические симптомы (парезы, атаксия и др.).

Клиническая картина Непереносимость физических нагрузок, после которых ухудшается самочувствие, появляется мышечная слабость, иногда миалгии. Инсультоподобные эпизоды проявляются рецидивирующими приступами головной боли, головокружением, развитием очаговой неврологической симптоматики (парезы, параличи конечностей, черепных нервов), коматозное состояние. Судороги при синдроме MELAS очень вариабельны-фокальные пароксизмы, генерализованные тонико-клонические, миоклонии. Судороги мало чувствительны к противосудорожных терапии.

Клиническая картина Деменция, обычно, развивается вместе с прогрессированием заболевания, но относительно редко она выступает в роли манифестного симптома. Миопатический симптомокомплекс (мышечная слабость, быстрая утомляемость, иногда гипотрофия). При раннем дебюте заболевания его ход более злокачественный. Так, при дебюте MELAS до 20 лет летальность составляет 30%.

Основные диагностические критерии: непереносимость физических нагрузок;начало заболевания до 40 лет (чаще до 20 лет);инсультоподибные эпизоды;судороги;"рваные" красные волокна в биоптатах скелетных мышц; лактат-ацидоз;прогрессирующая деменция;миопатический синдром;низкорослость;глухота.

Дополнительные диагностические критерии: кальцификация базальных ганглиев при компьютерной томографии (КТ) или магнитно - резонансной томографии (МРТ) головного мозга;атаксия;коматозные состояния;атрофия зрительных нервов;пигментный ретинит;синдром Вольфа - Паркинсона – Уайта;сердечная недостаточность;прогрессирующая наружная офтальмоплегия;нарушение проводимости сердца;сахарный диабет.

Данные лабораторных и функциональных исследований: Характерным признаком заболевания является выявление лактатацидоза в крови и спинномозговой жидкости. У половины больных в ликворе выявляется повышение уровня лактата и белка.Большое значение имеет исследование ферментов дыхательной цепи, чаще выявляются изменения в активности ферментов комплекса I. ЭКГ: могут выявляться нарушения сердечной проводимости, синдром Вольфа-Паркинсона-Уайта. КТ головного мозга: зоны инфарктов чаще в гемисферы, реже в мозжечке, базальных ганглиях. Может наблюдаться кальцинация базальных ганглиев, атрофия коры головного мозга. Церебральная ангиография: увеличение калибра сосудов (артерий, вен, капилляров).

МРТ головного мозга больной А., Т2взвешенные изображения. а, б – 1е исследование: симметричные очаги повышенной интенсивности сигнала в проекции теменных долей обоих полушарий. в, г – 2е исследование: в проекции височной и теменной долей правого полушария отмечается расширение зоны измененного МР сигнала. В левом полушарии в проекции теменной до ли размеры патологического очага заметно уменьшились.

КТ головного мозга: в височной доле левого полушария с частичным распространением на теменную долю определяется очаг слабо пониженной плотности (стрелка). В обоих полушариях в области лентикулярных ядер и зрительных бугров определяются очаги повышенной плотности (кальцификаты).

Исследование церебральной перфузии методом ОФЭКТ у больной А. а–в – исследование 12.11.2003 г. – зона низкой перфузии (синий и зеленый цвета) в височной области правого полушария (а) и высокой перфузии (красный цвет) в затылочных долях (б) и височной доле левого полушария (в). г–е –исследование 18.02.2004 г. – снижение перфузии в левой височной доле (е) по сравнению с предыдущим исследованием.

ДНК-диагностика: Поиск точечных мутаций в митохондриальной ДНК, характерных для синдрома MELAS. В большинстве случаев (80-90%) обнаруживается мутация в нуклеотиде 3243, где происходит замена аденина на гуанин в гене транспортной РНК для лейцина. Мутации характерные для синдрома MELAS могут проявляться у родственников пробанда при малосимптомно и асимптомно вариантах. Для того чтобы возникли клинические проявления синдрома MELAS, клон клеток с мутантным геном должен составлять от 56-до 95%.

Лечение Симптоматическое.Для коррекции биохимических дефектов используется коэнзим Q10 (80 - 300 мг / сут), витамины К1, К3 - филлохинон (25 мг / сутки) и менадион (до 75 мг / сут), янтарная кислота (до 6 мг / сут), витамин С (2-4 г / сут) и другие витамины (рибофлавин, тиамин, никотинамид). Известно, что коэнзим Q10 в физиологических условиях переносит электроны от комплексов I и II к комплексу III и содействует тем самым стабилизации дыхательной цепи, уменьшению уровня лактата и пирувата. Витамины К1 и К3, очевидно, способны выполнять функцию транспорта электронов на уровне I и III комплексов. Янтарная кислота обеспечивает передачу электронов II комплекса. Витамин С рассматривается как донор электронов IV комплекса, а также как важный антиоксидант. Кроме аскорбиновой кислоты, для предупреждения кислородно - радикального повреждения митохондриальных мембран назначается витамин Е (300 - 500 мг / сутки).

Лечение С целью стимуляции синтеза АТФ предлагается использовать идебенон (90 - 180 мг / сут), который обладает свойством усиливать энергетический метаболизм в мозговой ткани. Введение витаминов рибофлавина (100 мг / сутки) и никотинамида (до 1 г / сут) - предшественников коэнзимов НАД и ФАД, принимающих активное участие в окислительных процессах, также способствуют улучшению энергетической продукции митохондрий. В связи со вторичным карнитиновим дефицитом, больным назначают L-карнитин (до 100 мг / сутки). С целью снижения уровня лактата в крови и спинно-мозговой жидкости используется дихлорацетата натрия (25-100 мкг / кг).С помощью лабораторных тестов нужно проверить возможные нарушения функций эндокринной системы (сахарный диабет, гипопаратиреоз) и сердечно-сосудистой системы (блокада). При выявлении нарушений проводится их медикаментозная коррекция.

Прогноз Для жизни и выздоровления неблагоприятный.Лечение недостаточно эффективно.

Дифференциальный диагноз Другие митохондриальные заболевания (Синдром Кернса-Сейр, MERRF, синдром Лея (подострая некротизирующий энцефалопатия)), сахарный диабет, гипопаратиреоз, сердечные блокады, антифосфолипидный синдром, дефицит карнитина. Значительные трудности вызывает дифференцировку синдрома MELAS с такими нозологическим формам, как синдромы Кернса - Сейр и MERRF, особенно в начальной стадии болезни.

Благодарю за внимание!











