



, структурні аномалії X-хромосоми: Відзначають також мозаїчні варіанти хромосомних аномалій (45XO/46XX, 45XO/46XY), структурні аномалії X-хромосоми: ізохромосома Х (Хi), …")
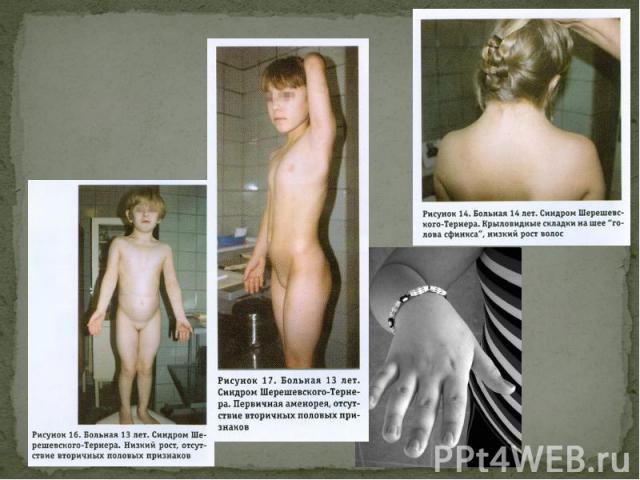
, низькорослість (98%), Загальна диспластичність (неправильна тілобудова) (92%), Діжкоподібна грудна клітка(75%), Вкорочення шиї (63%), Низький ріст волосся на шиї(57%), високе «готичне» піднебіння (56%), Крилоподібні складки шкі…")
, Деформація вушних раковин (46%), Вкорочення метакарпальних і метатарзальних кістой і аплазія фаланг (46%), Деформація ліктьових суглобів (36%), Множинні пігментніе родимки (35%), Лімфостаз (24%), Пороки серця і вилик…")
 і маса тіла 2800—2500 г менше). Для доношених новорождених характерні мала довжина (42—48 см) і маса тіла 2800—2500 г менше). Для перебігу постнатального періоду характерно загальний несп…")




 Соматотропін (можна досягти збільшення кінцевого росту пацієнта на 4-6 см); 1) Соматотропін (можна досягти збільшення кінцевого росту пацієнта на 4-6 см); 2) Анаболічні стероїди (мінімально ефективними дозами з перервами при регулярному гінеколог…")




 Пренатальна ( УЗД і лабораторні) Збільшення комірцевого простору Багатоводдя Гідроторакс У новонародженого відзначається затимка росту (довжина при народженні 48см і менше) при нормальній мас…")



Презентация на тему: Синдром Нунам і Шереревського-Тернера


Вперше ця хвороба як спадкова була описана в 1925 р. Н.О. Шерешевским, який вважав, що вона обумовлена недоразвиненням статевих жалоз, передньої долі гіпофиза і поєднується з вродженими дефектами розвитку Вперше ця хвороба як спадкова була описана в 1925 р. Н.О. Шерешевским, який вважав, що вона обумовлена недоразвиненням статевих жалоз, передньої долі гіпофиза і поєднується з вродженими дефектами розвитку

В 1938 р. Тернер виділив характерну для цього симптомокомплексу тріаду симптомів: статевий інфантилізм, шкірні крилоподібні складки на бічних поверхнях шиї і деформацію ліктьових суглобів. В 1938 р. Тернер виділив характерну для цього симптомокомплексу тріаду симптомів: статевий інфантилізм, шкірні крилоподібні складки на бічних поверхнях шиї і деформацію ліктьових суглобів. Етіологія захворювання (моносомія по Х-хромосомі) була досліджена Ч.Фордом в 1959 р.
,")
Відзначають також мозаїчні варіанти хромосомних аномалій (45XO/46XX, 45XO/46XY), структурні аномалії X-хромосоми: Відзначають також мозаїчні варіанти хромосомних аномалій (45XO/46XX, 45XO/46XY), структурні аномалії X-хромосоми: ізохромосома Х (Хi), Кільцева Х-хромосома (rX), делеція короткого плеча Х-хромосоми (Хр-) делеція довгого плеча Х-хромосоми (Хq-).
, низькорослість (98%), Загальна диспластичність (неправильн")
низькорослість (98%), низькорослість (98%), Загальна диспластичність (неправильна тілобудова) (92%), Діжкоподібна грудна клітка(75%), Вкорочення шиї (63%), Низький ріст волосся на шиї(57%), високе «готичне» піднебіння (56%), Крилоподібні складки шкіри в ділянці шиї (46%)
, Деформація вушних раковин (46%), Вкорочення мет")
Деформація вушних раковин (46%), Деформація вушних раковин (46%), Вкорочення метакарпальних і метатарзальних кістой і аплазія фаланг (46%), Деформація ліктьових суглобів (36%), Множинні пігментніе родимки (35%), Лімфостаз (24%), Пороки серця і виликих соглобів (22%), Підвишений артеріальний тиск (17%).
 і маса тіла 2800—2")
Для доношених новорождених характерні мала довжина (42—48 см) і маса тіла 2800—2500 г менше). Для доношених новорождених характерні мала довжина (42—48 см) і маса тіла 2800—2500 г менше). Для перебігу постнатального періоду характерно загальний неспокій новорождених, порушення смоктального рефлекса, зригування фонтаном, рвота. В ранньому віці у частини хворих відзначають затримку статичного розвитку і розвитку мови, що свідчить про патологію ембріогенезу нервової системи.

Молочні залози у більшості хворих не розвинені, соски розміщені низько. Молочні залози у більшості хворих не розвинені, соски розміщені низько. Вторинне оволосіння появляєть раптово, слабовиражене. Матка недорозвинена. Статеві залози не розвинені і представлені переважно сполучною тканиною. Геродермія (патологічна атрофія шкіри, нагадує старечу) Мошонкоподібний вигляд великих статевих губ Недорозвинення малих статевих губ, дівочої плівчи і клітора Воронкоподібний вхід у піхву.


Патологічне синостозірування відмічаєтся в метаепіфізарних зонах скелета, поодинокі чи множеинні аномалії розвитку кісток — частіше всього в променевозапястних суглобах, кістках кистей, колінних суглобах і хребті. На рентгенограмі кистей і променевозап’ясткового суглоба відзначається відставання кісткового віку від паспортного,затримка формування скелета зазвичай на 3—3,5 роки)

Визначення статевого хроматина Визначення статевого хроматина Дослідження каріотипу. УЗД дослідження звизначенням шийно-комірцевії ділянки

 Соматотропін (можна досягти збільшення кінцевого росту пацієнта на 4-6 см); 1")
1) Соматотропін (можна досягти збільшення кінцевого росту пацієнта на 4-6 см); 1) Соматотропін (можна досягти збільшення кінцевого росту пацієнта на 4-6 см); 2) Анаболічні стероїди (мінімально ефективними дозами з перервами при регулярному гінекологічному контролі); 3) Естрогени (після досягнення віку 12-13 років) 4) Кальцій-містять препарати

Для життя сприятливий, виключення складають хворі з важкими вродженими вадами серця і великих судин, з ренальної гіпертензією. Лікування естрогенами робить хворих здатними до сімейного життя, проте абсолютна більшість з них залишаються безплідними. Для життя сприятливий, виключення складають хворі з важкими вродженими вадами серця і великих судин, з ренальної гіпертензією. Лікування естрогенами робить хворих здатними до сімейного життя, проте абсолютна більшість з них залишаються безплідними. Хворі можуть успішно вчитися і виконувати будь-яку роботу, не пов'язану не пов'язану з фізичним і значним нервово-психічним напруженням.


Спадкове захворювання успадковується за аутосомно-домінантним й аутосомно-рецисивним типом, рідкісна вроджена патологія, носить сімейний характер, однаково зустрічається в хлопчиків та дівчаток. В 50% випадків можлива молекулярно генетична верифікація мутацій гена PTPN11. Є фенокопією хвороби Шерешевського-Тернера Спадкове захворювання успадковується за аутосомно-домінантним й аутосомно-рецисивним типом, рідкісна вроджена патологія, носить сімейний характер, однаково зустрічається в хлопчиків та дівчаток. В 50% випадків можлива молекулярно генетична верифікація мутацій гена PTPN11. Є фенокопією хвороби Шерешевського-Тернера

Жаклін Нунан практикуюча в якості кардіолога-педіатра дослідивши сполучення клінічної картини вродженого пороку серця з іншими аномаліями розвитку на прикладі 833 пацієнтів, в 1962р. написала статтю: «Поєднання несерцевих аномалій у дітей з уродженим пороком серця», в якій описала 9 дітей, на фоні вродженого пороку серця, що мали характерні риси обличчя, деформації грудної клітки, невеликий зріст. Патологія зустлічалась як в чоловіків, так і в жінок, при цьому кількість хромосом залишається нормальним. Жаклін Нунан практикуюча в якості кардіолога-педіатра дослідивши сполучення клінічної картини вродженого пороку серця з іншими аномаліями розвитку на прикладі 833 пацієнтів, в 1962р. написала статтю: «Поєднання несерцевих аномалій у дітей з уродженим пороком серця», в якій описала 9 дітей, на фоні вродженого пороку серця, що мали характерні риси обличчя, деформації грудної клітки, невеликий зріст. Патологія зустлічалась як в чоловіків, так і в жінок, при цьому кількість хромосом залишається нормальним.
 Пренатальна ( УЗД і лаборат")
Пренатальна ( УЗД і лабораторні) Пренатальна ( УЗД і лабораторні) Збільшення комірцевого простору Багатоводдя Гідроторакс У новонародженого відзначається затимка росту (довжина при народженні 48см і менше) при нормальній масі тіла. З народження діагностується клапанний стеноз легеневої артерії, крипторхізм у хлопчиків в 60% випадків, комбінована деформація грудної клітки.
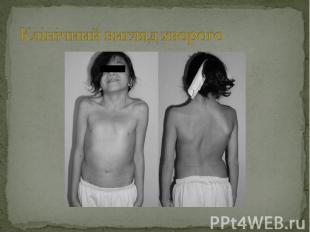

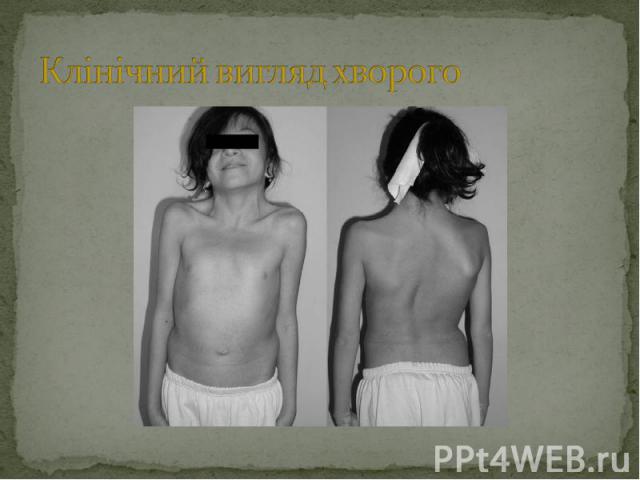
Головні клінічні прояви мають схожість з синдромом Шерешевского-Тернера: Головні клінічні прояви мають схожість з синдромом Шерешевского-Тернера: криловидні складки на шиї, вальгусна деформація ліктевих суглобів, низькорослість, лімфатичні набряки кистей і стоп; Інші прояви синдрому : птоз, запала грудна клітка, вроджені пороки правої половини серця (стеноз легеневої артерії), трикутне обличчя і розумова відсталість. У хлопчиків відзначають порушення розвитку яєчок (крипторхізм, атрофія, анорхія, зменшення просвіту сім’євивідних канальців зі склерозом або без нього, зменшення або відсутрість гермінтативних клітин, гіперплазія клетин Лейдинга). Деякі пацієнти з нормальними яєчками зберігають фертильність, але в більшості відмічають помірний або виражений гіпогонадизм. Вміст тестостерону в плазмі низький, або визначаєть на нижніх межах норми, рівень гонадотропіну підвищений. Каріотип XY (нормальний, чоловічий). Причина затримки росту не уточнена, так як рівень базального і стимульованого гормона росту нормальний.


Симптоматичне. За наявності ознак гіпогонадизму лікування полягає в замісній терапії препаратами статевих гормонів. Застосування гормону росту неефективне. За показами проводиться хірургічна корекція вроджених вад розвитку, лікування психічних порушень, пов'язаних із затримкою розумового розвитку. Симптоматичне. За наявності ознак гіпогонадизму лікування полягає в замісній терапії препаратами статевих гормонів. Застосування гормону росту неефективне. За показами проводиться хірургічна корекція вроджених вад розвитку, лікування психічних порушень, пов'язаних із затримкою розумового розвитку.












