






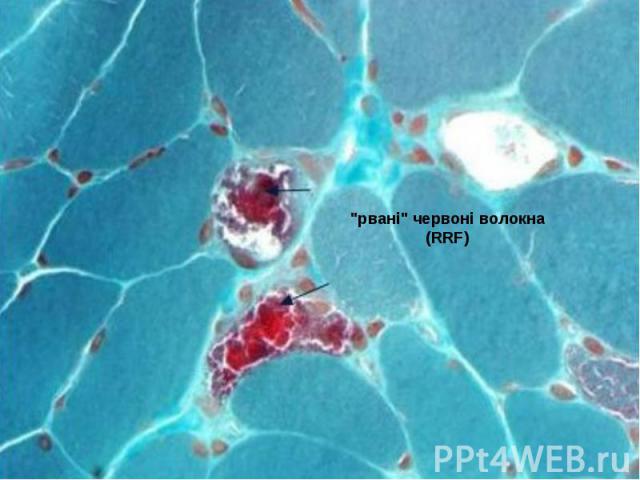
, являются \"рвані\" червоні волокна (RRF), которые проявляються в м’язовій тканині при модифіко…")


 мозочкову атаксію і прогресуючу м'язову слабкість міоклонії зазвичай провокуються звуковими …")




, хворобами накопичування (наприклад, хвороба Краббе), синдромом Айкарді та іншими дизгенезіями …")

Презентация на тему: Синдром MERRF



Синдром MERRF перше описаний N.Fukuhara із співробітниками в 1980 році.

Синдрому MERRF серед дорослої популяції в Фінляндії складає 1,5:100 000, на півночі Англії - 0,25: 100 000 і 0,25:100 000 серед дитячого населення на заході Швеції. Синдрому MERRF серед дорослої популяції в Фінляндії складає 1,5:100 000, на півночі Англії - 0,25: 100 000 і 0,25:100 000 серед дитячого населення на заході Швеції.

Рідкісне мітохондріальне захворювання. Рідкісне мітохондріальне захворювання. Викликане мутаціями в наступних генах: MTTK, MTTL1, MTTN, MTTS1, MTTS2, MTTF. Симптоми MERRF також проявляються при мутаціях гена MTND5. Обумовлене точковими мутаціями в гені тРНК-лізин у позиціях 8344 і 8356 мтДНК.

Зазвичай синдром MERRF успадковується по материнській лінії, за винятком нових мутацій, які вперше виникли в сім’ї. Зазвичай синдром MERRF успадковується по материнській лінії, за винятком нових мутацій, які вперше виникли в сім’ї.

Характерним патоморфологічним признаком синдрома MERRF, як і ряду інших мітохондріальних енцефаломіопатій (синдромів Кернса - Сейр, MELAS та ін.), являются "рвані" червоні волокна (RRF), которые проявляються в м’язовій тканині при модифікованому фарбуванні трихромом за Гоморі. Вони являются морфологічим субстратом пошкодження мітохондріальної ДНК і утворються внаслідок проліферації аномальних мітохондрій. Характерним патоморфологічним признаком синдрома MERRF, як і ряду інших мітохондріальних енцефаломіопатій (синдромів Кернса - Сейр, MELAS та ін.), являются "рвані" червоні волокна (RRF), которые проявляються в м’язовій тканині при модифікованому фарбуванні трихромом за Гоморі. Вони являются морфологічим субстратом пошкодження мітохондріальної ДНК і утворються внаслідок проліферації аномальних мітохондрій. Червоні "рвані" волокна являються наслідком мутацій, пошкоджуючих гени транспортної РНК і приводять до порушень внутрішньомітохондріального синтезу білка.

А – феномен «рваних» червоних волокон. Б – вигляд здорового м’язу. А – феномен «рваних» червоних волокон. Б – вигляд здорового м’язу. Фарбування за Гоморі трихромом, збільшення х400.

Вік початку захворювання - від юнацького до п'ятого десятиліття життя. Ранній вік початку корелює з тяжкістю клінічних проявів. Клінічний фенотип часто представлений неповними клінічними формами і характеризується вираженим внутріродинним клінічним поліморфізмом. Вік початку захворювання - від юнацького до п'ятого десятиліття життя. Ранній вік початку корелює з тяжкістю клінічних проявів. Клінічний фенотип часто представлений неповними клінічними формами і характеризується вираженим внутріродинним клінічним поліморфізмом. Також клінічна картина захворювання в уражених родичів по материнській лінії може варіювати від практично безсимптомної до м'яко вираженої, включаючи тільки нейросенсорну туговухість та затримку росту.

Основний симптомокомплекс Основний симптомокомплекс включає: різні види епілептичних нападів (міоклонічні, генералізовані тоніко-клонічні, атонічні та інші) мозочкову атаксію і прогресуючу м'язову слабкість міоклонії зазвичай провокуються звуковими подразниками і фотостимуляцією нейросенсорна туговухість поліневропатичний синдром зниження інтелекту атрофія зорових нервів спастичні парези / паралічі і ліпоми Пігментна дегенерація сітківки, хронічний панкреатит та цукровий діабет зустрічаються рідко.

материнський тип успадкування; материнський тип успадкування; дебют у віці 3-65 років; ураження ЦНС: міоклонус, атаксія, деменція у поєднанні з нейросенсорною глухотою, атрофією зорових нервів, порушенням глибокої чутливості; лактат – ацидоз; помірне підвищення білка в лікворі; недостатність I, III, IV комплексів дихального ланцюга; ЕЕГ: генералізованні комплекси “ спайк-хвиля”; ЕМГ: первинно-м'язовий тип ураження; КТ: атрофія мозку, лейкоенцефалопатія, іноді кальцифікація базальних гангліїв; “рвані червоні волокна” в біоптатах кістякових м’язів; прогресуючий перебіг.

Насамперед, спрямоване на: Насамперед, спрямоване на: корекцію порушень енергетичного обміну зменшення лактат-ацидозу попередження ушкоджень мембран мітохондрій вільними радикалами. Встановлено ефективність рибофлавіну, нікотинаміду, цитохрому С та коензиму Q10, L-карнітину, вітаміну С.

Велике значення має також протисудомна терапія. Препаратами першої черги є: вальпроати по 30мг/кг/добу, а при їх неефективності – клоназепам.

Залежить від тяжкості генетичного дефекту. У випадках, коли пул мутантної мітохондріальної ДНК в тканинах перевищує 85-90%, прогноз абсолютно несприятливий, і фатальний результат настає через 5-10 років від моменту появи перших симптомів. У інших хворих можлива тривала компенсація стану на тлі протисудомної терапії і застосування препаратів, що сприяють корекції лактат-ацидозу і стабілізації окислювально-відновного комплексу мітохондрій. Залежить від тяжкості генетичного дефекту. У випадках, коли пул мутантної мітохондріальної ДНК в тканинах перевищує 85-90%, прогноз абсолютно несприятливий, і фатальний результат настає через 5-10 років від моменту появи перших симптомів. У інших хворих можлива тривала компенсація стану на тлі протисудомної терапії і застосування препаратів, що сприяють корекції лактат-ацидозу і стабілізації окислювально-відновного комплексу мітохондрій.

Проводиться з іншими прогресуючими міоклонус-епілепсіями (хворобою Гоше, галактосіалідозом ІІ типу, синдромом міоклонуса з нирковою недостатністю та ін.), хворобами накопичування (наприклад, хвороба Краббе), синдромом Айкарді та іншими дизгенезіями мозку. Проводиться з іншими прогресуючими міоклонус-епілепсіями (хворобою Гоше, галактосіалідозом ІІ типу, синдромом міоклонуса з нирковою недостатністю та ін.), хворобами накопичування (наприклад, хвороба Краббе), синдромом Айкарді та іншими дизгенезіями мозку. Значні труднощі викликає диференціювання синдрому MERRF з такими нозологічними формами, як синдроми Кернса - Сейр і MELAS, особливо в початковій стадії хвороби.












