


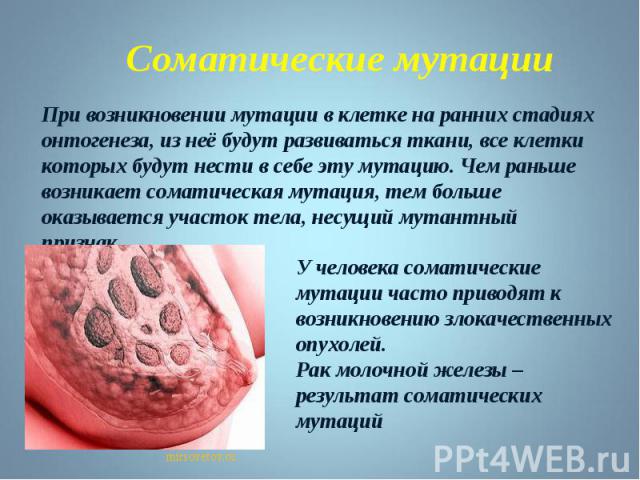


; Группа генетических болезней, возникающих в результате мутаций в соматических клетках (генетические соматические болезн…")



 мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана, боле…")








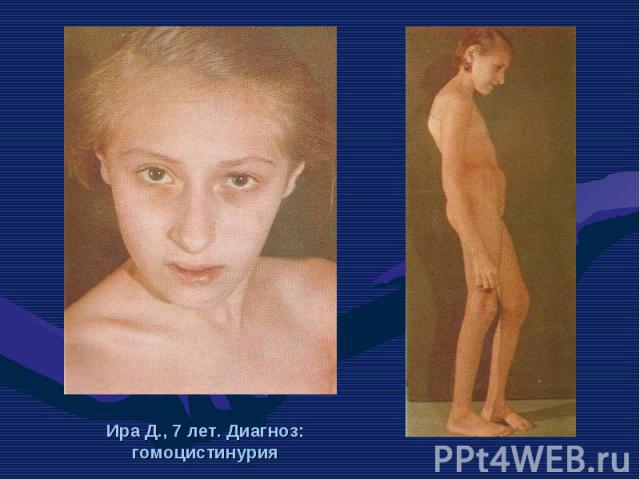
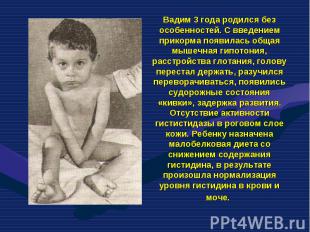
 – это она из самых частых форм наследственных дефектов обмена аминокислот. Фенилкетонурия (ФКУ) – это она из самых частых форм наследственных дефектов обмена аминокислот. Частота в европейских странах составляет 1:10 000 новорож…")


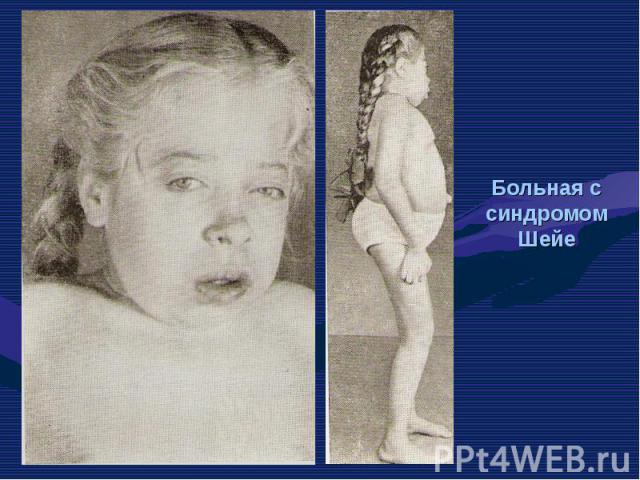
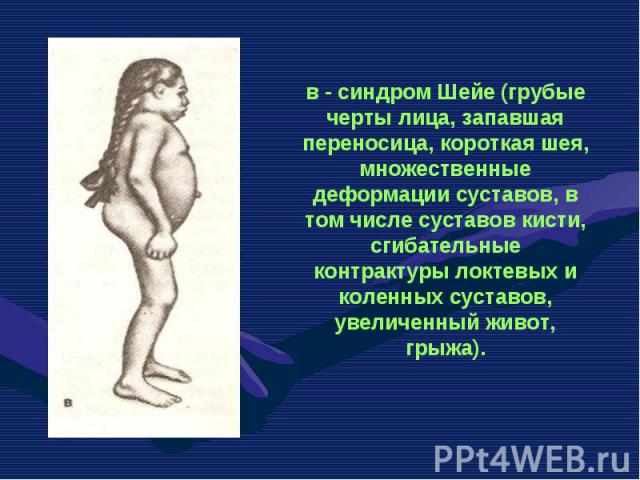
 остеогенеза (OI-I; …")

















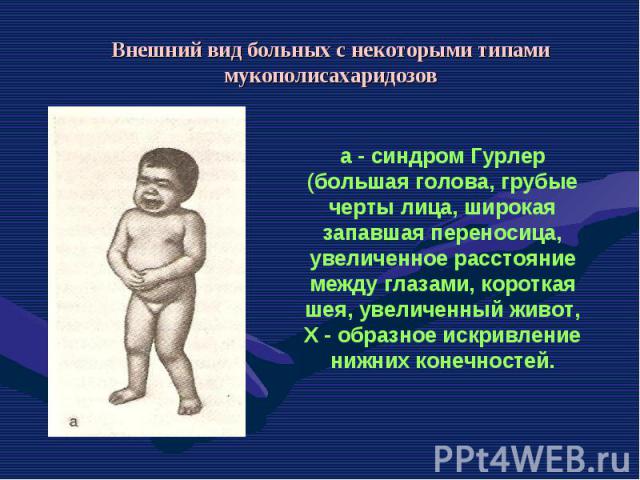


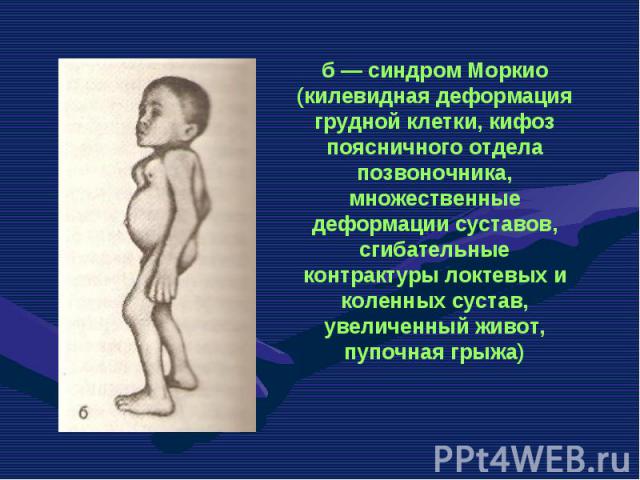


 — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным старением организма. Основными формами является детская прогерия (синдр…")




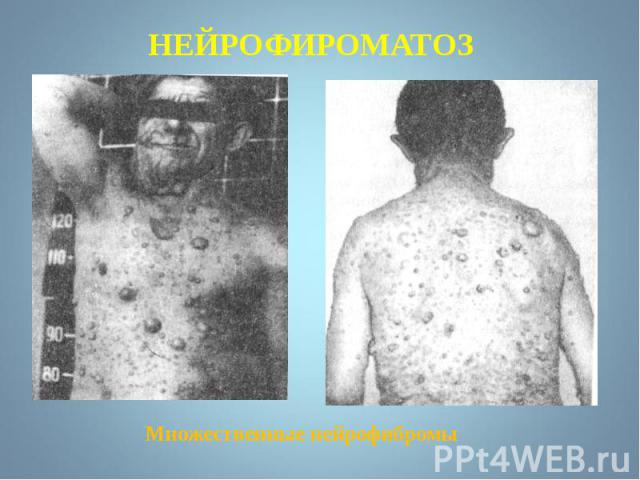
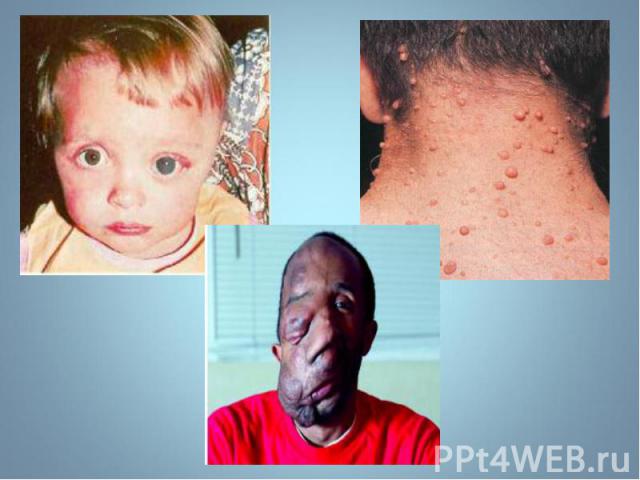
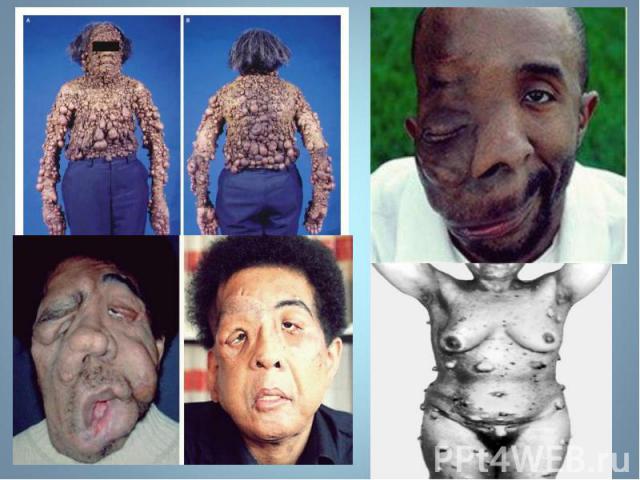
 - аутосомно-доминантное заболевание, характеризующееся наличием множественных пигментных пятен , кожных и подкожных опухолей . обнаруживаются небольшие гамартомы . Чаще наблюдается н…")




 относится к группе наследственных нарушений биосинтеза стероидных гормонов. Адреногенитальный синдром (АГС) относится к группе наследственных нарушений биосинтеза стероидных гормонов. Заболевание впервые описано в 186…")















Презентация на тему: Наследственные болезни человека





Генные болезни; Генные болезни; Хромосомные болезни; Болезни с наследственной предрасположенностью (мультифакториальные болезни); Группа генетических болезней, возникающих в результате мутаций в соматических клетках (генетические соматические болезни); Болезни генетической несовместимости матери и плода.

Ранняя манифестация; Ранняя манифестация; Хроническое прогредиентное течение; Относительная резистентность к терапии; Множественность поражения; Семейный характер заболевания; Клинический полиморфизм.

Генные болезни - это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне. Число известных в настоящее время моногенных наследственных заболеваний составляет около 4000 нозологических форм. Встречаются эти заболевания с частотой 1:500 – 1:100 000 и реже.

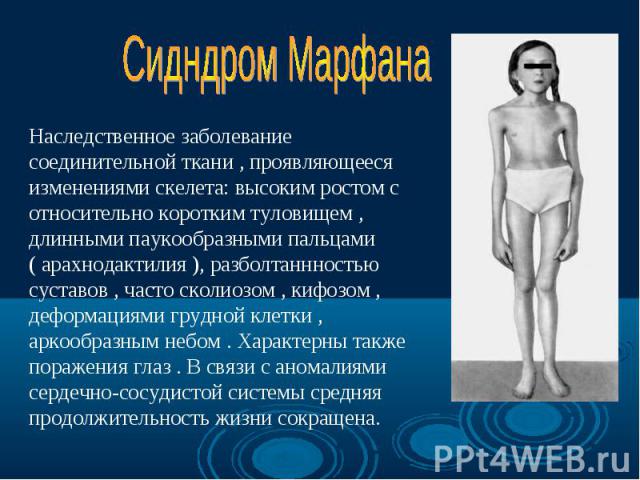
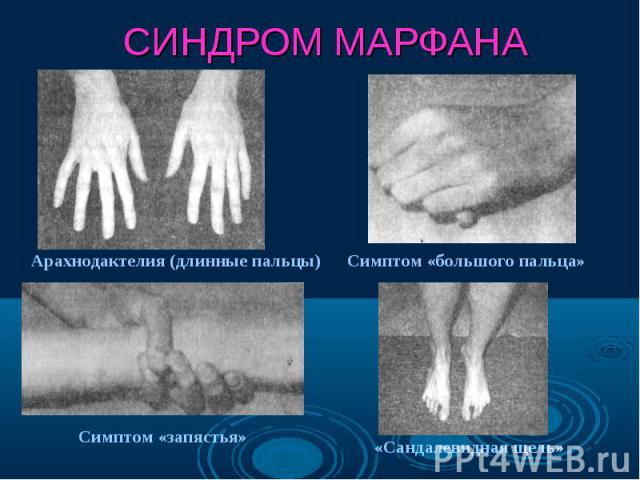
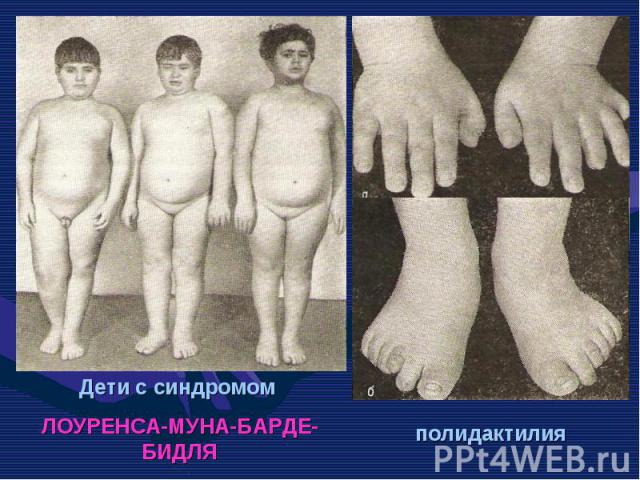
Нейрофиброматоз Нейрофиброматоз Синдром Марфана Болезнь Олбрайта Дизостозы Отосклероз Пароксизмальная миоплегия Талассемия Семейная гипехолестеринемия Несовершенный остеогенез

мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана, болезни Гунтера, болезни Фабри (рецессивное наследование, сцепленное с Х хромосомой) мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана, болезни Гунтера, болезни Фабри (рецессивное наследование, сцепленное с Х хромосомой) фосфат-диабет (доминантное наследование, сцепленное с Х хромосомой)
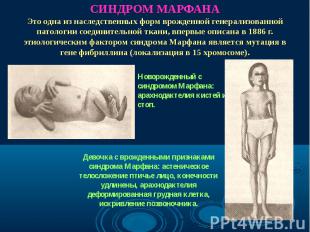

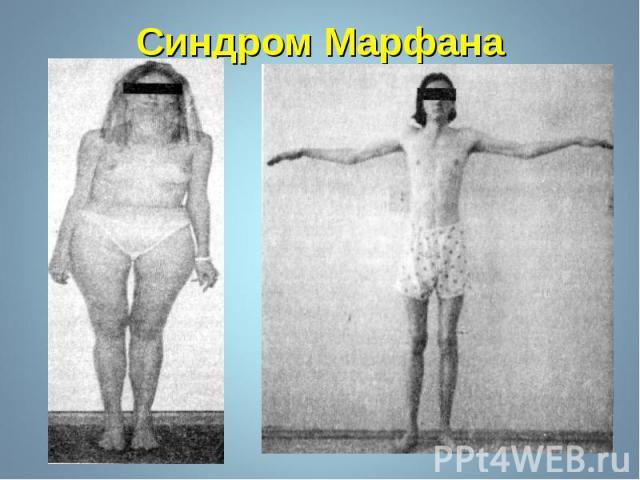
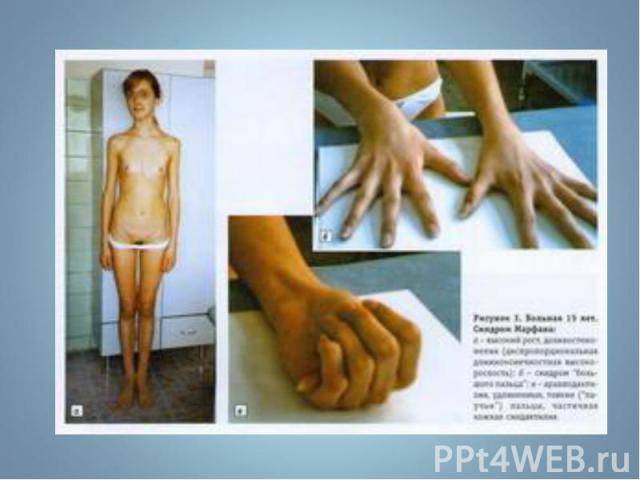
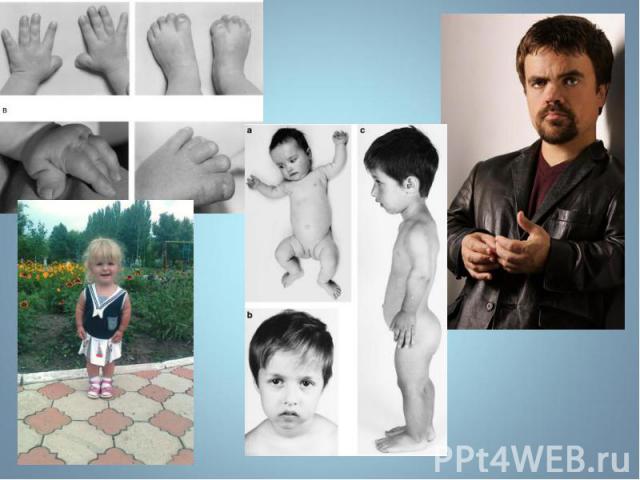
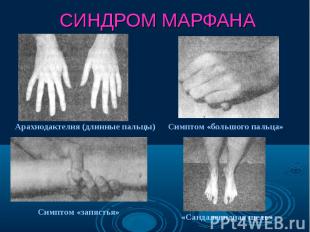
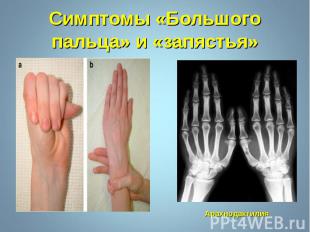
Синдром Марфана - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем, длинными паукообразными пальцами, разболтанностью суставов, часто сколиозом, кифозом, деформациями грудной клетки, аркообразным небом. Синдром Марфана - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем, длинными паукообразными пальцами, разболтанностью суставов, часто сколиозом, кифозом, деформациями грудной клетки, аркообразным небом.

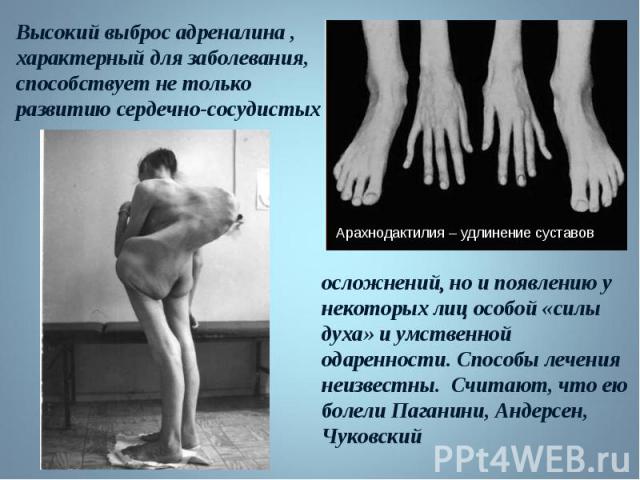
Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны. Считают, что ею болели Паганини, Андерсен, Чуковский. Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны. Считают, что ею болели Паганини, Андерсен, Чуковский.



Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечнососудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечнососудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны

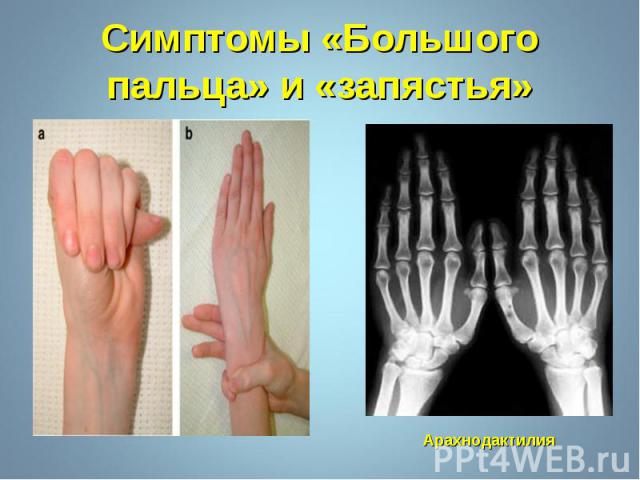
Впервые описан в 1896 г. Впервые описан в 1896 г. Клинические признаки: высокий рост, арахнодактилия, подвывих хрусталика, порок митрального клапана, плоскостопие, гипоплазия мышц. Тип наследования – АД Частота наследования – 0,04 : 1000.

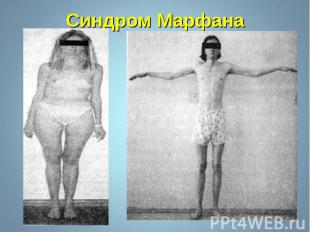


Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация – повышенное содержание адреналина в крови, поэтому больные всю жизнь находятся в возбужденном состоянии и становятся невероятными трудоголиками. Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация – повышенное содержание адреналина в крови, поэтому больные всю жизнь находятся в возбужденном состоянии и становятся невероятными трудоголиками. Синдромом Марфана страдали всемирно известные личности: Авраам Линкольн – президент США (рост 193 см), Ганс Христиан Андерсен – великий писатель,

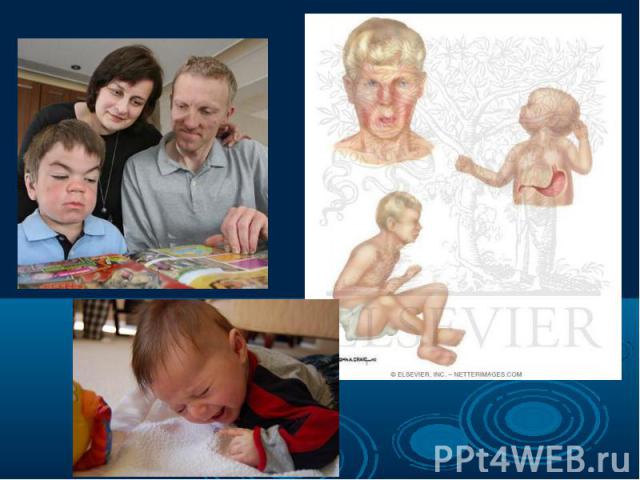
 – это она из самых частых форм наследственных дефектов обме")
Фенилкетонурия (ФКУ) – это она из самых частых форм наследственных дефектов обмена аминокислот. Фенилкетонурия (ФКУ) – это она из самых частых форм наследственных дефектов обмена аминокислот. Частота в европейских странах составляет 1:10 000 новорожденных. В Турции частота составляет 1:2600, в Ирландии – 1:4500, в Швеции – 1:30 000, в Японии – 1:119 000. В основе ФКУ лежит дефицит фенилаланин – 4 – гидроксилазы, фермента, контролирующего превращение фенилаланина в тирозин. Аутосомно – рецесивный ген, кодирующий данный фермент, картирован на 12 – й хромосоме.


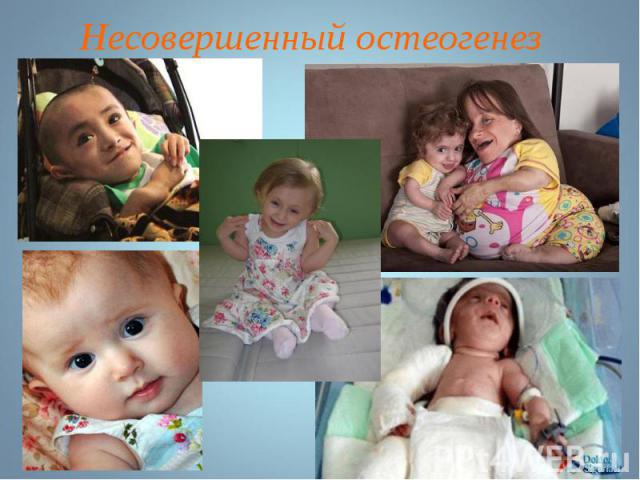
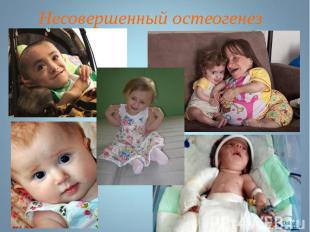
Несовершенный остеогенез наследуется по аутосомно-рецессивному типу. Несовершенный остеогенез наследуется по аутосомно-рецессивному типу.

Несовершенный остеогенез фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах, глухотой, голубыми склерами, аномалиями зубов . Клиницисты различают несколько типов (I-IV) остеогенеза (OI-I; OI-II; OI-III; OI-IV). Несовершенный остеогенез фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах, глухотой, голубыми склерами, аномалиями зубов . Клиницисты различают несколько типов (I-IV) остеогенеза (OI-I; OI-II; OI-III; OI-IV).

Ахондроплазия — врожденное поражение скелета — врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется карликовостью, короткими конечностями при обычной длине туловища, деформацией нижних конечностей и позвоночника и относительной макроцефалией. Ахондроплазия — врожденное поражение скелета — врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется карликовостью, короткими конечностями при обычной длине туловища, деформацией нижних конечностей и позвоночника и относительной макроцефалией.

Человек, закончивших свой рост, достигает 30 - 41 см. Человек, закончивших свой рост, достигает 30 - 41 см.

Причины мутации в настоящее время не известны. Причины мутации в настоящее время не известны.




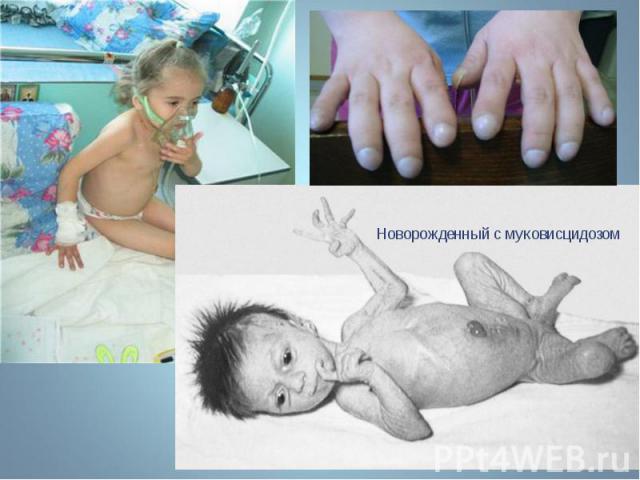
Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую очередь, поражением ЖКТ и органов дыхания. Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую очередь, поражением ЖКТ и органов дыхания.

Заболевание обусловлено генерализованным поражением экзокринных желез. Заболевание обусловлено генерализованным поражением экзокринных желез. Частота муковисцедоза среди новорожденных в европейской популяции составляет 1:2500. В то же время муковисцидоз редко встречается в восточных популяциях и у африканского черного населения (1:100 000). Ген муковисцидоза локализован на 7 – й хромосоме. В нем обнаружено около 1000 мутаций, примерно 300 из которых вызывают клинические проявления. Этот ген детерминирует синтез белка, называемого муковисцидозным трансмембранным регулятором проводимости.

Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую очередь, поражением ЖКТ и органов дыхания. Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую очередь, поражением ЖКТ и органов дыхания.
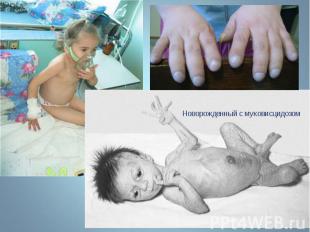

Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве, хотя в 4% случаев диагноз ставят в зрелом возрасте. Муковисцидоз наследуется аутосомно-рецессивно. Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве, хотя в 4% случаев диагноз ставят в зрелом возрасте. Муковисцидоз наследуется аутосомно-рецессивно.

Распространенность заболевания сильно отличается в разных этнических группах. Среди белого населения Северной Америки и Северной Европы муковисцидоз встречается у 1 из 3000 живых новорожденных, в то время как у американских негров - у 1 из 17000, а у полинезийцев, населяющих Гавайи, - лишь у 1 из 90000. Распространенность заболевания сильно отличается в разных этнических группах. Среди белого населения Северной Америки и Северной Европы муковисцидоз встречается у 1 из 3000 живых новорожденных, в то время как у американских негров - у 1 из 17000, а у полинезийцев, населяющих Гавайи, - лишь у 1 из 90000.








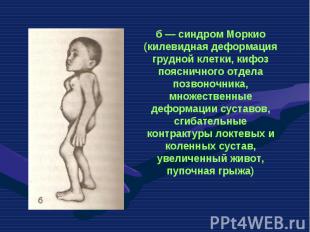
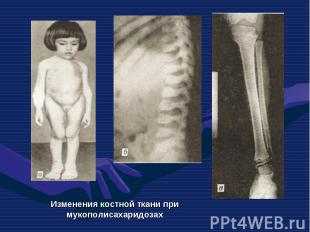

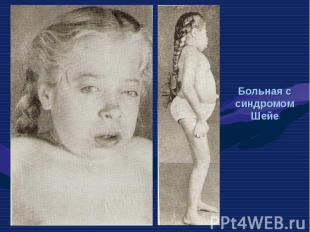
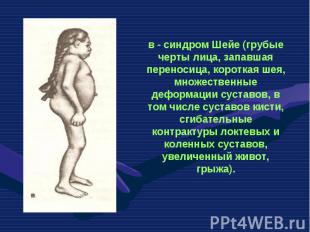
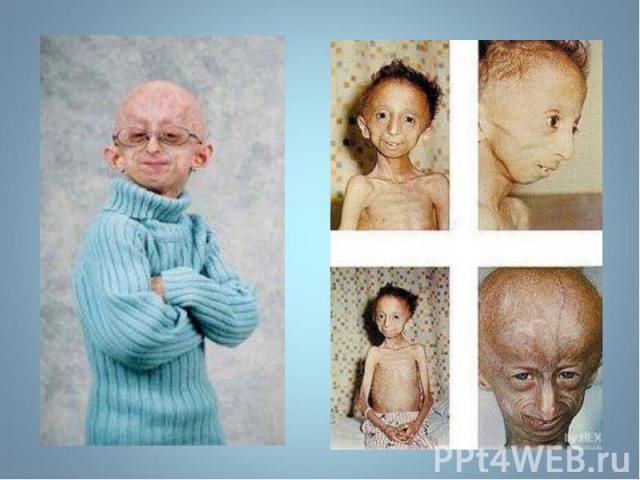
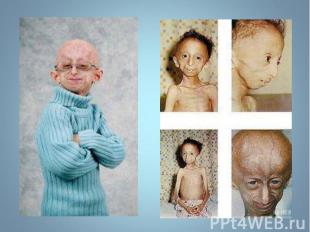
 — патологическое состояни")
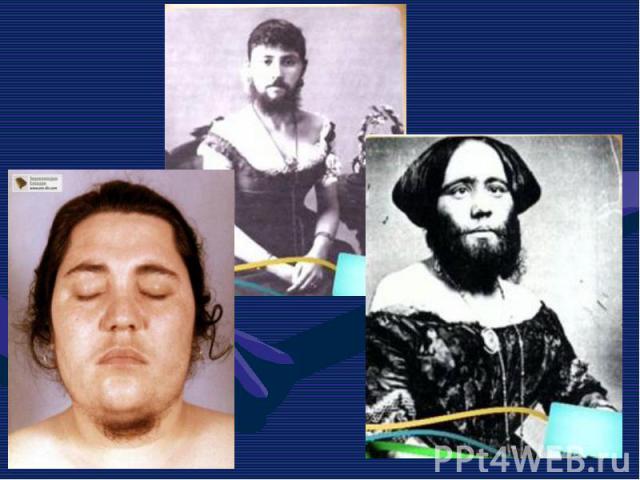
Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным старением организма. Основными формами является детская прогерия (синдром Гетчинсона (Хадчинсона) — Гилфорда) и прогерия взрослых (синдром Вернера). Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным старением организма. Основными формами является детская прогерия (синдром Гетчинсона (Хадчинсона) — Гилфорда) и прогерия взрослых (синдром Вернера).

Описана в 1886 г. Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз. Дети умирают в 13-15 лет после нескольких инфарктов и инсультов дряхлыми стариками. Болезнь вызывает мутантный ген LMNA, отвечающий за синтез белков Lamin A,B,C, необходимых для соединительной ткани. Наступает тотальная алопеция, на коже черепа выражена венозная сеть. Тип наследования и популяционная частота неизвестны


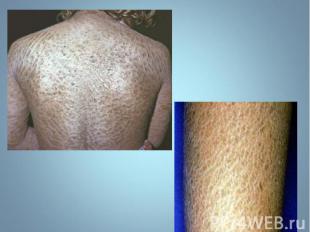


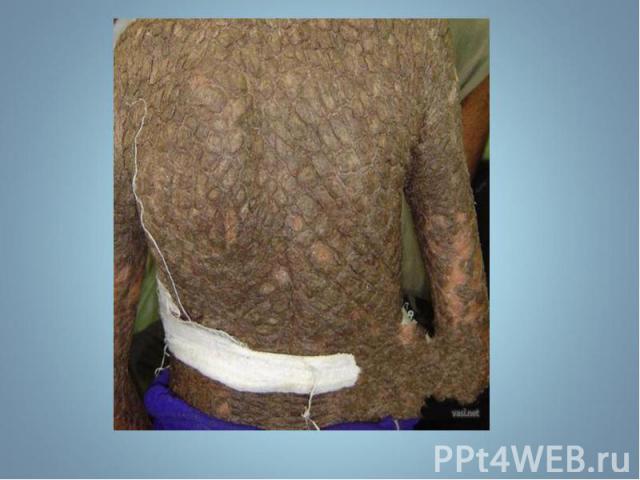
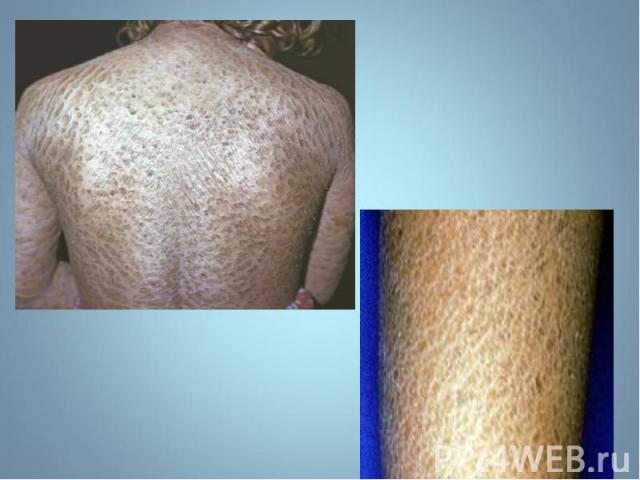
Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по размеру и окраске, часто кофе с молоком. Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по размеру и окраске, часто кофе с молоком.
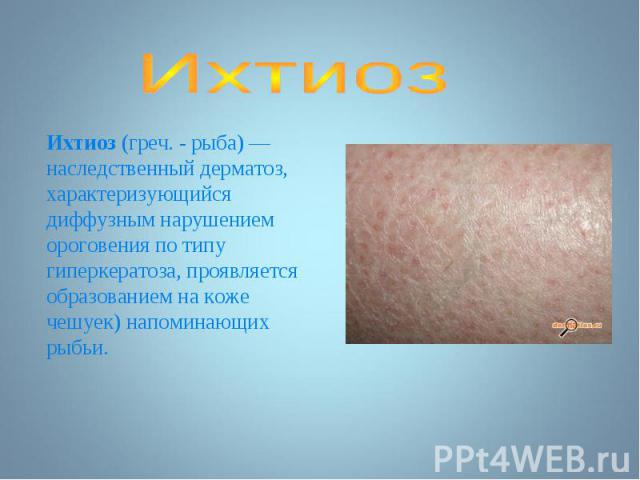
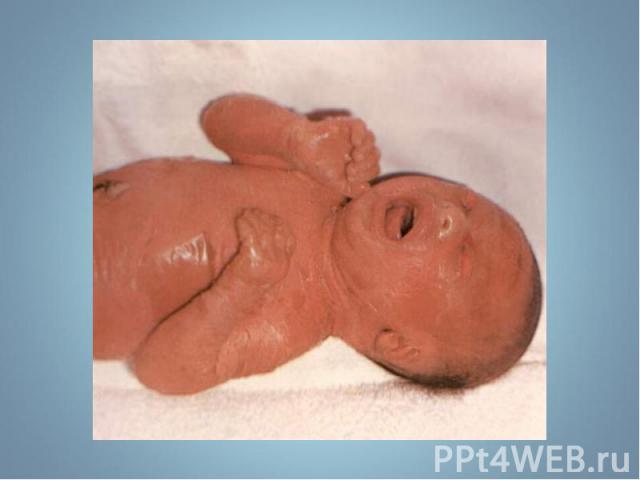
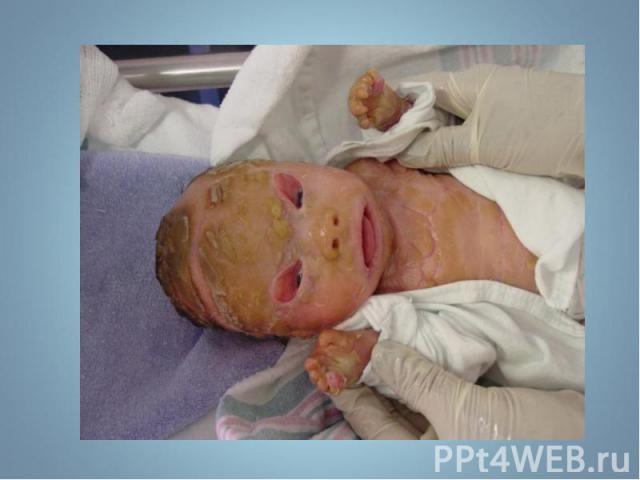
 - аутосомно-д")
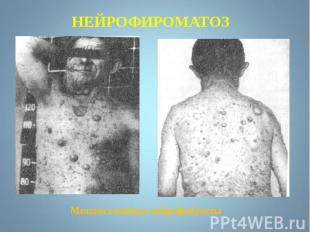
Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное заболевание, характеризующееся наличием множественных пигментных пятен , кожных и подкожных опухолей . обнаруживаются небольшие гамартомы . Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное заболевание, характеризующееся наличием множественных пигментных пятен , кожных и подкожных опухолей . обнаруживаются небольшие гамартомы .


Почти у всех больных на радужке обнаруживаются небольшие гамартомы . Нейрофибромы могут подвергаться злокачественной трансформации с развитием нейрофибросарком . Почти у всех больных на радужке обнаруживаются небольшие гамартомы . Нейрофибромы могут подвергаться злокачественной трансформации с развитием нейрофибросарком .

Мутация, за развитие этого типа, обнаружена в хромосоме 17, в зоне, кодирующей белок нейрофибромин, подавляющий рост опухолей. Мутация, за развитие этого типа, обнаружена в хромосоме 17, в зоне, кодирующей белок нейрофибромин, подавляющий рост опухолей.
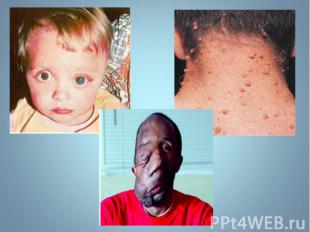
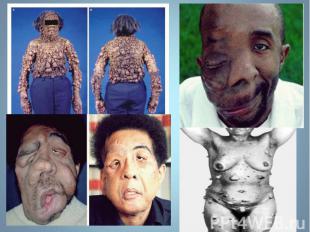

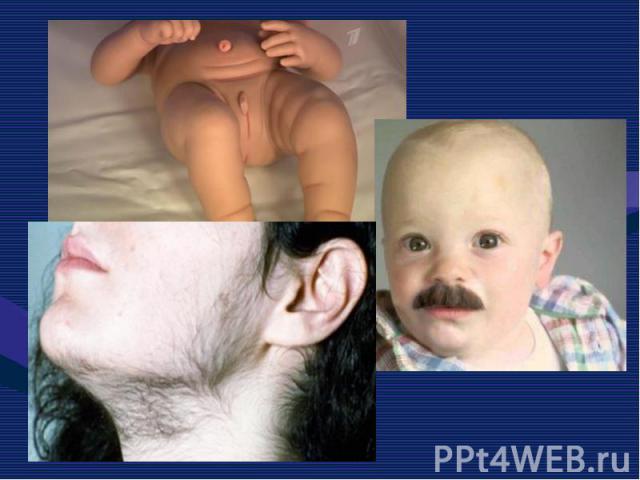
 относится к группе наследственных нарушений биос")
Адреногенитальный синдром (АГС) относится к группе наследственных нарушений биосинтеза стероидных гормонов. Адреногенитальный синдром (АГС) относится к группе наследственных нарушений биосинтеза стероидных гормонов. Заболевание впервые описано в 1865 г. Наиболее часто встречающуюся в 90% случаев форму – дефицит 21 – гидроксилазы.

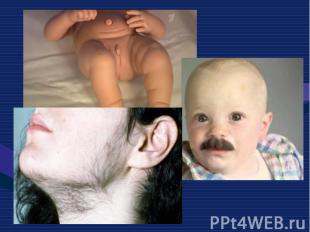


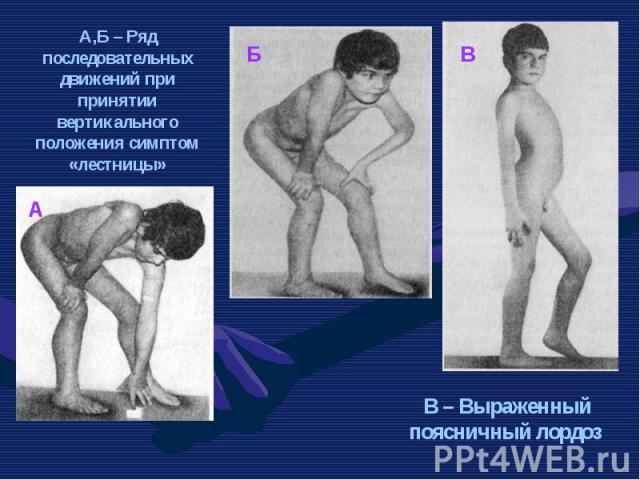
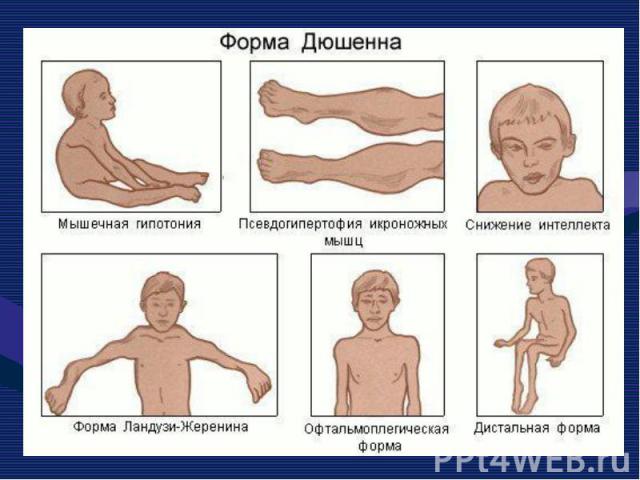
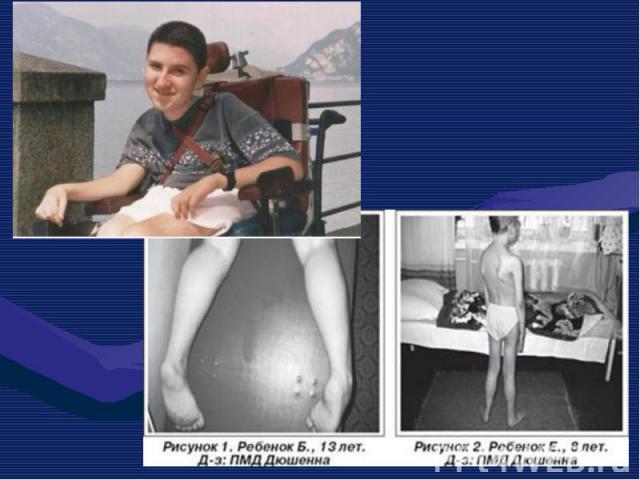
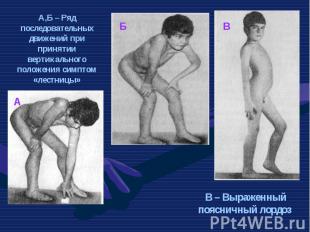
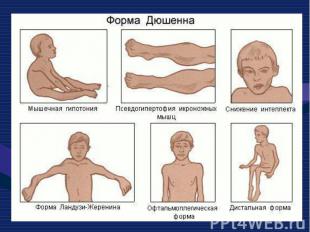
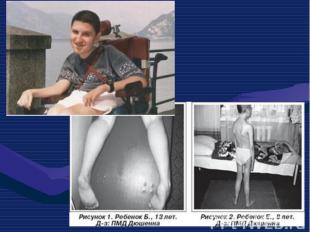
Это одна из самых частых форм наследственных нервно – мышечных заболеваний. Это одна из самых частых форм наследственных нервно – мышечных заболеваний. Впервые она была описана в 1868 г. Мышечные дистрофии характеризуются дегенеративными изменениями в поперечно – полосатой мускулатуре без первичной патологии периферического мотонейрона. Частота ее составляет 1:3000 – 1:5000 мальчиков.

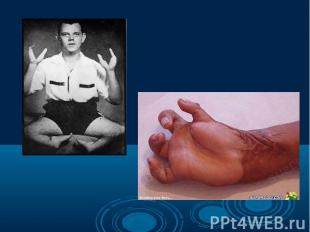

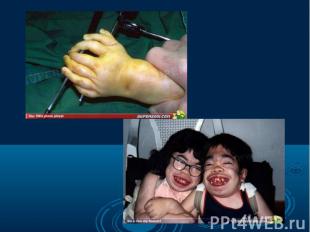
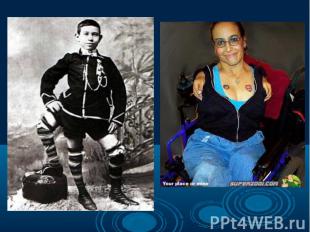


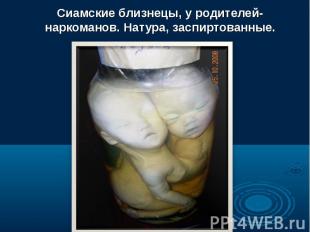





Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов мутаций одном и том же гене, которые являются причиной наследственных болезней. Даже одна и та же генная болезнь может быть обусловлена разными мутациями. Например, в гене муковисцидоза описано свыше 200 вызывающих болезнь мутаций. Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов мутаций одном и том же гене, которые являются причиной наследственных болезней. Даже одна и та же генная болезнь может быть обусловлена разными мутациями. Например, в гене муковисцидоза описано свыше 200 вызывающих болезнь мутаций.

Если принять, что у человека примерно Если принять, что у человека примерно 100 000 генов и каждый ген может мутировать и контролировать синтез белка с другим строением, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в различных участках гена).

Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации происходят постоянно как отрицательные, так и положительные. Положительные мутации позволяют приспосабливаться к новым условиям окружающей среды и мира и ведут к дальнейшим мутациям. Отрицательные же виды мутаций не поддерживаются окружающей средой и человеком и не могут носить дальнейшего развития. Однако надо учитывать вероятность того, что отрицательные ранее мутации могут повториться с человеком вновь и при новых обстоятельствах окружающей среды и человека могут приносить новые результаты мутаций. Какими они будут покажет будущее. Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации происходят постоянно как отрицательные, так и положительные. Положительные мутации позволяют приспосабливаться к новым условиям окружающей среды и мира и ведут к дальнейшим мутациям. Отрицательные же виды мутаций не поддерживаются окружающей средой и человеком и не могут носить дальнейшего развития. Однако надо учитывать вероятность того, что отрицательные ранее мутации могут повториться с человеком вновь и при новых обстоятельствах окружающей среды и человека могут приносить новые результаты мутаций. Какими они будут покажет будущее.












