

 — сложный патофизиологический процесс зарождения и развития опухоли. (син. онкогенез). Выделяют следующие стадии формирования опухоли Гиперплазия ткани Добр…")


. При различных онкологических заболеваниях регистрируется значительное повышение активности этих генов (рак поджелудочной железы, рак мочевого пузыря и т.д.). …")


 ДНК протекает с большой точностью благодаря функци…")


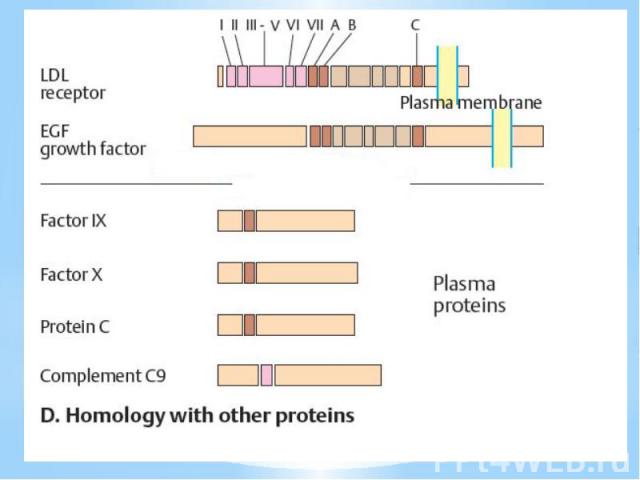


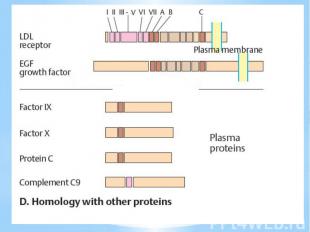
 приводит к развитию карциномы мочевого пузыря; точковая мутация в…")



, эти гены «…")




Презентация на тему: Генетические аспекты онкогенеза

Генетические аспекты онкогенеза Студентки 4 курса Датиевой И.А.

Канцерогене з (лат. cancerogenesis; cancer — рак + др.-греч. γένεσις — зарождение, развитие) — сложный патофизиологический процесс зарождения и развития опухоли. (син. онкогенез). Выделяют следующие стадии формирования опухоли Гиперплазия ткани Доброкачественная опухоль Дисплазия Рак in situ Инвазивный рак Вторая стадия (формирование доброкачественной опухоли) может отсутствовать. Рак in situ прорастает базальную мембрану. Опухолевые клетки разрушают и замещают собой предсуществующий эпителий. В дальнейшем раковые клетки врастают в лимфатические и кровеносные сосуды с последующим переносом опухолевых клеток и образованием метастазов. Прорастание опухолевых клеток через базальную мембрану и инвазия в подлежащую ткань. Врастание в кровеносные и лимфатические сосуды

Генетические аспекты этиологии и патогенеза злокачественных новообразований у человека изучаются очень давно. Первым предположил роль наследственности, как одного из механизмов канцерогенеза, французский хирург Брока, описавший в 1869 г. родословную семьи своей жены, где из 24 женщин 10 умерли от рака молочной железы. Как и для всех мультифакториальных заболеваний реализация механизмов канцерогенеза обеспечивается совместным действием целого ряда генетических и средовых факторов. Основными генетическими механизмами, запускающими процесс канцерогенеза, у человека являются мутации генов двух групп семейств, контролирующих процессы жизнеобеспечения клетки: протоонкогенов и антионкогенов (генов-супрессоров опухолевого роста).

Протоонкогены — это группа нормальных генов клетки, оказывающая стимулирующее влияние на процессы клеточного деления, посредством специфических белков — продуктов их экспрессии. Превращение протоонкогена в онкоген (ген – определяющий опухолевые свойства клеток) является одним из механизмов возникновения опухолевых клеток. Это может произойти в результате мутации генетического кода протоонкогена с изменением структуры специфического белка продукта экспрессии гена, либо же повышением уровня экспрессии протоонкогена при мутации его регулирующей последовательности (точечная мутация) или при переносе гена в активно транскрибируемую область хромосомы (хромосомные аберрации). Хромосомный набор нормальной (слева) и раковой клетки

На данный момент изучена канцерогенная активность протоонкогенов группы ras (HRAS, KRAS2). При различных онкологических заболеваниях регистрируется значительное повышение активности этих генов (рак поджелудочной железы, рак мочевого пузыря и т.д.). Так же раскрыт патогенез лимфомы Беркитта активация протоонкогена MYC происходит в случае его переноса в область хромосом где содержатся активно транскрибируемые коды иммуноглобулинов.

Гены-супрессоры представлены группой генов, чья функция противоположна функции протоонкогенов. Гены-супрессоры оказывают тормозящее влияние на процессы клеточного деления и выхода из процесса дифференцировки. Доказано, что в ряде случаев инактивация генов-супрессоров с исчезновением их антагонистического влияния по отношению к протоонкогенам ведет к развитию некоторых онкологических заболеваний. Так, потеря участка хромосомы, содержащего гены-супрессоры, ведет к развитию таких заболеваний, как ретинобластома, опухоль Вильмса и др.

Таким образом, система протоонкогенов и генов-супрессоров формирует сложный механизм контроля темпов клеточного деления, роста и дифференциации. Нарушения этого механизма возможны как под влиянием факторов внешней среды, так и в связи с геномной нестабильностью — теория, предложенная Кристофом Лингауром и Бертом Фогельштейном. Питер Дюсберг из Калифорнийского университета в Беркли утверждает, что причиной опухолевой трансформации клетки может быть анеуплодия (изменение числа хромосом или потеря их участков), являющаяся фактором повышенной нестабильности генома.

По мнению некоторых ученых, еще одной причиной возникновения опухолей мог бы быть врожденный или приобретенный дефект систем репарации клеточной ДНК. В здоровых клетках процесс репликации (удвоения) ДНК протекает с большой точностью благодаря функционированию специальной системы исправления пострепликационных ошибок. В геноме человека изучено по крайней мере 6 генов, участвующих в репарации ДНК. Повреждение этих генов влечет за собой нарушение функции всей репаративной системы, и, следовательно, значительное увеличение уровня пострепликационных ошибок, то есть мутаций (Lawrence A. Loeb).


Протоонкогены представляют собой группу семейств генов, которые играют ключевую роль в пролиферации и дифференцировке клеток, функционировании клеточных рецепторов, репарации ДНК и формировании ответа на внешние регуляторные сигналы. Продукты этих генов в норме регулируют многостадийный процесс сигнальной трансдукции. Передача сигналов опосредована тремя механизмами:

Трансформация нормальной клетки в опухолевую представляет собой длинную цепь событий, инициируемых каскадом мутаций в протоонкогенах и генах-супрессорах. Механизмы канцерогенеза, запускаемые мутациями в генах этих двух групп семейств, несколько различаются. Мутации в протоонкогенах, обусловливающие их трансформацию в так называемые клеточные (целлюлярные) онкогены (с-опс), могут

Мутации в структурной части гена - это, как правило, точковые мутации, меняющие конфигурацию белка, в результате чего активируется аутофосфорилирование, увеличивается активность фермента и, следовательно, усиливается клеточный рост. Эти мутации аутосомно-доминантные, так как для злокачественной трансформации клетки не нужен второй сопутствующий онкоген.

Такие мутации описаны для семейства протоонкогенов ras: так в гене c-Ha-ras в кодоне, соответствующем 12-й аминокислоте белка p21ras, замена G -> Т (в белке - замена глицина на валин) приводит к развитию карциномы мочевого пузыря; точковая мутация в гене c-N-ras, приводящая к замене лизина на глицин в 61-м положении белка р21ras, обусловливает развитие меланом и ряда карцином; активация протонкогена c-Ki-ras2 вследствие двух различных точковых мутаций в одном и том же кодоне, соответствующем 12-й аминокислоте белка р21 ras (замена глицина на валин или цистеин), приводит к развитию острого миелоидного лейкоза или карциномы щитовидной железы, соответственно. Точковые мутации выявлены и в гене erbA: замена ряда аминокислот в аминотерминальных положениях на валин в белке P75erb приводит к развитию эритробластозов.

При анализе кариотипа больных с различными типами опухолей часто выявляются транслокации, делеции и инсерции. В результате этих хромосомных перестроек протоонкоген может попасть в зону действия активного промотора, при этом транскрипция протоонкогена увеличится. Так протоонкогены c-mos, с-тус и с-аbl активируются вследствие транслокаций t(8;21), t(8;14), t(9;22), соответственно, что приводит к развитию следующих опухолей: острого миелоидного лейкоза, лимфомы Беркитта, хронического миелоидного лейкоза. Делеции 11р рядом с областью расположения гена c-ras - Пр13, могут привести к развитию аниридии или опухоли Вилмса.

Результатом транслокации может быть и амплификация гена, например, амплификация протонкогена аЫ при хроническом миелолейкозе. Предполагается, что инсерции в регуляторные участки гена представлены преимущественно последовательностями генома некоторых ДНК-содержащих вирусов или провирусной ДНК ретровирусов (РНК-содержащие). Встраивание в определенные участки генома регуляторных ДНК-последовательностей ретровирусов и/или активация провирусами промоторов клеточных онкогенов может привести к резкому усилению экспрессии протоонкогенов и развитию опухоли.

Возникновение опухолей, ассоциированных с ДНК-содержащими вирусами, обусловлено тем, что вирусная ДНК становится частью клеточного генома и вместе с ним передается дочерним клеткам. При этом репликация и экспрессия вирусной ДНК частично или полностью регулируются клеточными механизмами, в зависимости от степени интеграции генома вируса.

Предполагают, что онкогены ретровирусов имеют клеточную природу и возникли вследствие рекомбинации провирусной ДНК с генами, регулировавшими процессы деления, дифференцировки и метаболизма клетки. Становясь вирусными (лишенными интронов), эти гены «уходят» от клеточного контроля регуляции экспрессии, постоянно функционируют и амплифицируются. Следствием этих событий является их повышенная экспрессия, приводящая к трансформации нормальной клетки в опухолевую. Гомология протоонкогенов с провирусными ДНК-последовательностями ретровирусов объясняет роль вирусов в некоторых возможных механизмах злокачественного перерождения клеток.

При рекомбинации и внедрении в геном вируса последовательности, соответствующей протоонкогену, вследствие высокого уровня мутирования ретровирусов, вероятность трансформации протоонкогена в онкоген возрастает. Далее процесс трансформации клетки может идти двумя путями: либо при последующей рекомбинации происходит обратное встраивание онкогена в клетку и экспрессия измененного белка, либо увеличение дозы аномального гена путем амплификации.

Гомология протоонкогенов и вирусных онкогенов лежит в основе и их обозначения. Для этого используется аббревиатура, состоящая из первых букв названия вируса, в котором впервые был обнаружен данный ген, а иногда индуцируемой им опухоли и поражаемого вида. Механизм амплификации гена имеется и в норме, гак как необходим для защиты клетки от стрессовых ситуаций. Например, резистентность опухолевых клеток к химиотерапии мегатрексатом при миелоидном лейкозе обусловлена амплификацией гена дигидрофолат-редуктазы - фермента, блокируемого этим препаратом. Наиболее часто наблюдается амплификация протоонкогенов тус-семейства. Этот механизм активации протоонкогена обнаружен при таких опухолях как нейробластома, около половины случаев которой обусловлено амплификацией гена N-тус, и мелкоклеточная карцинома легких, возникающая вследствие амплификации генов с-тус: N-myc и L-myc.



")

")
")


")



