


, связанная с неконтролируемой активацией системы комплемента . Основным органом-мишенью микроангиопатического тромбообразования служат почки, однако в ряде случаев возможна генерализация ТМА,…")



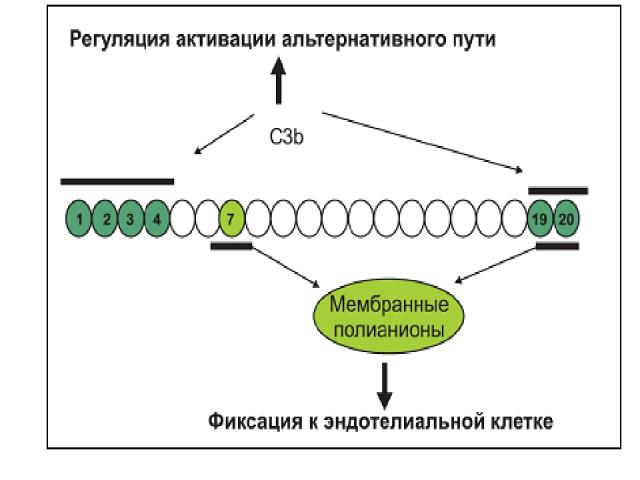
 является наиболее важным протеином в системе регуляции альтернативного пути активации комплемента . Он состоит из 20 коротких консенсусных повторов (SCRs) и содержит не менее двух C3b-связывающих участков. Первый участок, связывающийс…")
 встречаются крайне редко (1%), в то время как мутации C3 фракции комплемента встречаются у 10% пациентов. Приблизительно 10% детей имеют …")

 характерно для аГУС, связанного с мутациями CFH и CFI (средний возраст 6 мес и 2 мес — соответственно). Наоборот, при мутации MCP заболевание всегда начинается старш…")


, с наличием или отсутствием анурии, олигоурии, протеинурии при …")
, ацидоз (сывороточный бикарбонат")
, субэндотелиальным накоплением протеинов и клеточного детрита, фибринов…")


 отсутствие ассоциированного заболевания; 2) отсутствие критериев ГУС, связанного с Шига-токсином (посев кала и ПЦР на Шига-токсины; серология на антилипополисахаридные антитела); 3) отсутствие критери…")
 и антител к фактору Н, генетический скрининг факторов риска. За исключением определения плазменной концентрации C3 и…")
: у пациентов с началом заболевания в возрасте до одного года в первую очередь необходимо проводить скрининг CFH, CFI и C3, вне зависимости о…")




 и трансплантации почки, то и прогноз в значител…")
, человеческие моноклональные антитела к C5. Экулизумаб известен…")
 Для варианта с дефицитом HFI разрабатывается рекомбинантный HFI, а также возможность использования донорской плазмы, богатой HFI .")
Презентация на тему: ГУС

Атипический гемолитико-уремический синдром.

Атипичный вариант ГУС составляет 5-10% от всех случаев ГУС у детей, является генетическим прогрессирующим заболеванием с крайне высоким риском внезапной смерти и необратимых инвалидизирующих повреждений жизненно важных органов, включая почки, печень, сердце и мозг
, связанная с неконтроли")
В основе аГУС лежит тромботическая микроангиопатия (ТМА), связанная с неконтролируемой активацией системы комплемента . Основным органом-мишенью микроангиопатического тромбообразования служат почки, однако в ряде случаев возможна генерализация ТМА, приводящая к развитию полиорганной ишемии с картиной полиорганной недостаточности

Заболеваемость и распространенность аГУС изучены недостаточно. По данным различных источников, в странах с достаточным уровнем диагностики распространенность заболевания составляет от 1 до 3 пациентов аГУС на 1 миллион населения. Болезнь чаще манифестирует у детей до 18 лет, но приблизительно в 40% случаев диагностируется во взрослом возрасте.

Патогенез Еще в 1970-1980 гг. было замечено, что ряд пациентов с аГУС имели низкий уровень C3 плазмы крови. Впечатляющий прогресс был достигнут в течение последних 10 лет, продемонстрировавший, что 4 регуляторных белка альтернативного пути активации комплемента фактор H (CFH), мембранный кофакторный белок (MCP или CD46), фактор I (CFI) и тромбомодулин (THBD), а также 2 протеина С3-конвертазы, С3 и фактор B (CFB) играют роль в патогенезе аГУС
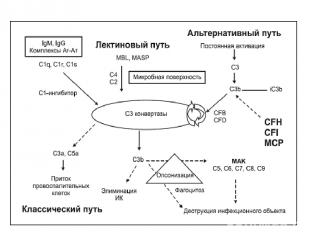

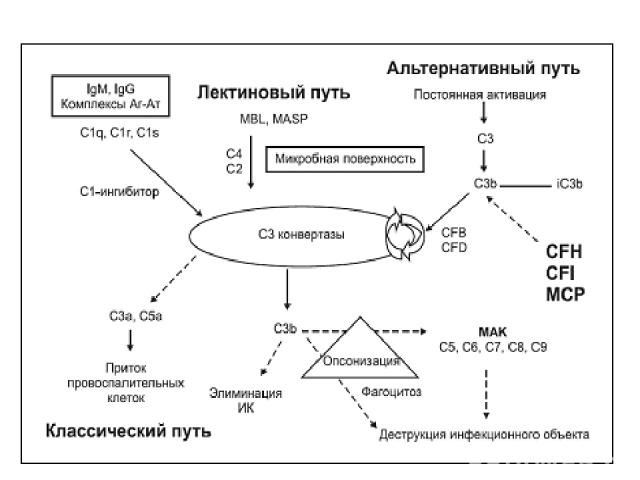
Все эти три пути сходятся в точке расщепления C3 фракции комплемента. В то время как активация классического и лектинового путей начинается после связывания с иммунными комплексами или микроорганизмами соответственно, альтернативный путь активируется постоянно с образованием C3b, который беспорядочно связывается с патогенами и клетками хозяина. На чужеродной поверхности, например бактериальной, C3b связывает СFB, который затем расщепляется фактором D с формированием C3-конвертазы C3bBb. C3bBb в геометрической прогрессии расщепляет C3 (петля усиления) и формирует С5-конвертазу (C3bBb (C3b)n). Компонент C5b, получаемый в результате расщепления С5, принимает участие в образовании мембраноатакующего комплекса (МАК) C5b9, который вызывает опсонизацию, фагоцитоз и лизис бактерий). Указанная реакция в норме строго контролируется на поверхности клеток хозяина, которые защищены от локальной амплификации депозитов C3b рядом контролирующих систему комплемента протеинов: CFH (гликопротеин плазмы, кофактор для CFI), CFI (сериновая протеаза плазмы, которая расщепляет и инактивирует C3b с формированием iC3b в присутствии кофакторов, включающих МСР (нециркулирующий гликопротеин, встроенный в мембраны всех клеток, кроме эритроцитов) и, возможно, THBD ( тромбомодулин). THBD – эндотелиальный гликопротеин, обладающий антикоагулянтными, противовоспалительными и цитопротективными свойствами, также являющийся регуляторным белком системы комплемента. В присутствии CFH отмечается конкуренция CFH и CFB за связывание C3b, что ограничивает образование С3-конвертазы. Когда CFH связан с C3b, закрепленным на поверхности клетки, CFB более не имеет возможности принимать участие в формировании С3-конвертазы
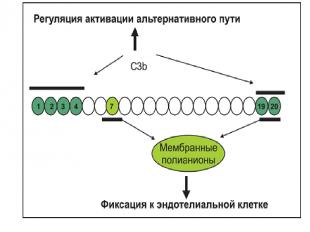
 является наиболее важным протеином в системе регуляции альтернати")
CFH (фактор H) является наиболее важным протеином в системе регуляции альтернативного пути активации комплемента . Он состоит из 20 коротких консенсусных повторов (SCRs) и содержит не менее двух C3b-связывающих участков. Первый участок, связывающийся с C3b, регулирующий жидкую фазу амплификации альтернативного пути, располагается в N-терминальном конце SCR1–4. Второй C3b-связывающий участок расположен в SCR19–20 С терминального домена. CFH также содержит два полианионных связывающих участка в SCR7 и SCR19–20. Эндотелиальные клетки богаты полианионными молекулами, в частности, гликозаминогликанами. Защита клеток хозяина зависит от инактивации поверхностно-связанного C3b, вторичной по отношению к связыванию CFH с поверхностно-связанным C3b. Все последние научные исследования наглядно продемонстрируют роль SCR19–20 в защите эндотелиальных клеток .Четыре протеина – CFH, CFI, MCP и THBD – локально взаимодействуют для расщепления C3b до неактивной молекулы (iC3b). Мутации этих белков приводят к утрате защиты эндотелиальных клеток

ГУС ассоциируется с мутациями CFH у 20-25% пациентов, MCP — у ≈15 % и CFI — у ≈10 %. Мутации фактора В (CFB) встречаются крайне редко (1%), в то время как мутации C3 фракции комплемента встречаются у 10% пациентов. Приблизительно 10% детей имеют сочетанные мутации. В дополнение, 10% детей имеют приобретенный функциональный дефицит CFH в связи с наличием анти-CFH антител. Только 30% aГУС сегодня не находит должного объяснения

Предполагается, что мутации в генах CFH, MCP, CFI и THBD, обнаруженные у пациентов с аГУС, приводят к образованию дефекта защиты эндотелиальных клеток от активации системы комплемента. В целом, все идентифицированные генетические дефекты приводят к усилению образования C3-конвертазы, а следовательно, и С5-конвертазы, и расщеплению C5. Вследствие этого усиливается высвобождение С5а и МАК на поверхности клеток эндотелия, вызывающее дополнительное их повреждение с обнажением субэндотелиального матрикса и образованием тромбов. Это приводит к потреблению тромбоцитов и повреждению эритроцитов. Кроме того, продемонстрирована значительная роль CFH в регуляции структуры и функции тромбоцитов. При наличии С-концевых мутаций способность CFH связываться с тромбоцитами снижается, что приводит к_активации комплемента на поверхности тромбоцитов. Это, в свою очередь, вызывает активацию тромбоцитов, их агрегацию, высвобождение тканевого фактора, экспрессирующего микрочастицы, и содействует образованию тромбов в микроциркуляторном русле

Клиническая манифестация аГУС. Очень раннее начало (даже в периоде новорожденности) характерно для аГУС, связанного с мутациями CFH и CFI (средний возраст 6 мес и 2 мес — соответственно). Наоборот, при мутации MCP заболевание всегда начинается старше 1 года. Этиологически неопределенные варианты аГУС могут начаться в любом возрасте. Анти-CFH антитела чаще отмечаются ближе к подростковому периоду. Провоцирующими факторами являются некоторые инфекции (верхних дыхательных путей, лихорадка, гастроэнтериты), они запускают первый эпизод аГУС и рецидивы независимо от генетического варианта у 2/3 пациентов. Диарея провоцирует аГУС у 1/3 пациентов, что иногда затрудняет дифференциацию с Д+ ГУС.

Приблизительно у 25% больных аГУС носит семейный характер. Неотягощенный семейный анамнез не исключает возможности генетической передачи заболевания. Лишь половина носителей мутации в семье в течение жизни имеют манифестацию заболевания.

Начало заболевания, как правило, внезапное Симптомами у детей раннего возраста являются бледность, общее недомогание, плохой аппетит, рвота, сонливость и иногда отеки Экстраренальные проявления в виде поражения ЦНС проявляются судорогами, сопором, комой и выявляются у менее чем 20% пациентов. В патогенезе энцефалопатии играют важную роль одновременно несколько факторов: отёк мозга, гипоксия, кровоизлияния в вещество мозга. Массивное внутрисосудистое потребление тромбоцитов может спровоцировать развитие коагулопатии потребления: гипер- или гипокоагуляция, гипопротромбинемия, гипофибриногенемия, повышение уровня продуктов деградации фибрина, Д-димера, замедленный фибринолиз, нарушение агрегации тромбоцитов

У большинства пациентов при первом лабораторном исследовании выявляется полная диагностическая триада ГУС: гемоглобин 40-90 г/л тромбоцитопения (8 ммоль/л, креатинин >110 мкмоль/л), с наличием или отсутствием анурии, олигоурии, протеинурии при сохраненном диурезе. Лейкоцитоз, умеренно выраженный ретикулоцитоз. Для подтверждения неиммунного характера анемии выполняется реакция Кумбса- отрицательная при ГУС. Наличие шизоцитов, а также неопределяемый гаптоглобин, в сочетании с высоким уровнем ЛДГ, подтверждают микроангиопатический внутрисосудистый генез гемолиза

В случае запоздалой диагностики могут наблюдаться опасные для жизни осложнения: гиперкалиемия (≥6 ммоль/л), ацидоз (сывороточный бикарбонат

При гистологическом анализе пораженных сосудов выявляется ТМА, характеризующаяся утолщением стенок артериол и капилляров, с выраженным повреждением эндотелия (отек и отслойка), субэндотелиальным накоплением протеинов и клеточного детрита, фибриновыми и тромбоцитарными тромбами, обтурирующими просвет сосуда. ТМА преимущественно поражает микрососуды почек, хотя в патологический процесс могут вовлекаться микрососуды головного мозга, сердца, легких и желудочно-кишечного тракта. В клинической картине могут присутствовать симптомы полиорганной недостаточности: поражение лёгких, поджелудочной железы, желудочно-кишечного тракта, миокарда.

В целом прогноз аГУС неблагоприятный. Смертность в острой стадии составляет 5-10%. Приблизительно у 50% пациентов развивается терминальная ХПН, чаще в течение 1 года от начала манифестации

Рецидивы аГУС отмечаются при всех вариантах, чаще у пациентов с мутацией MCP. Провоцирующие инфекции при этом сопровождаются острым гемолизом, тромбоцитопенией и острой почечной недостаточностью в результате гемоглобинурии. В большинстве этих случаев функция почек полностью восстанавливается. Промежуток времени между рецидивами может колебаться от нескольких недель до многих лет. Наиболее благоприятный прогноз отмечается при наличии MCP мутации, наиболее неблагоприятный — при CFH и сочетанных мутациях. По данным французских авторов, смерть или терминальная ХПН в течение менее чем 1 года от начала заболевания отмечены у 60% с мутацией CFH, у 37% с мутацией CFI, у 33% с мутацией С3, у 60% с комбинированными мутациями, у 32% в группе с неизвестной этиологией и 0% с мутацией MCP. У больных с анти-CFH антителами в случае раннего лечения ЗПП заболевание имеет благоприятное течение
 отсутствие ассоциированного заб")
Критериями постановки диагноза аГУС являются: 1) отсутствие ассоциированного заболевания; 2) отсутствие критериев ГУС, связанного с Шига-токсином (посев кала и ПЦР на Шига-токсины; серология на антилипополисахаридные антитела); 3) отсутствие критериев тромботической тромбоцитопенической пурпуры (активность ADAMTS13 сыворотки больше 10%). У детей ТТП чаще носит врожденный характер и связан с наследственным полным дефицитом ADAMTS13

Необходимо исследование системы комплемента С3, С4, концентрации в плазме фактора Н и фактора I, экспрессии на лейкоцитах MCP(СD46) и антител к фактору Н, генетический скрининг факторов риска. За исключением определения плазменной концентрации C3 и C4, исследования системы комплемента требуют наличия специализированных лабораторий [27]. При анализе родословных, по крайней мере, у 20% заболевание имеет наследственный характер с аутосомно-рецессивным или доминатным путем наследования.

У детей возраст начала указывает на необходимость выполнения конкретных генетических исследований (табл. 3 ): у пациентов с началом заболевания в возрасте до одного года в первую очередь необходимо проводить скрининг CFH, CFI и C3, вне зависимости от того, снижена концентрация C3 плазмы или нет. В случае начала заболевания в возрасте старше одного года и нормальной концентрации C3 требуется, в первую очередь, исключение мутации MCР. Учитывая тот факт, что анти-CFH-ГУС встречается, как правило, после семилетнего возраста и в раннем подростковом и подростковом периодах, приоритетным в этих возрастных группах является скрининг анти-CFH антител, особенно если снижена концентрация С3. Скрининг мутаций CFB и THBD необходим, если не выявлено мутаций CFH, CFI, MCP и С3, незвисимо от возраста начала заболевания

Лечение До настоящего времени терапией первой линии является плазмотерапия, при отсутствии бесспорных доказательств её эффективности СЗП является источником нормальных CFH, CFI, C3 и CFB, а также большого количества других функциональных белков. Плазмаобмен имеет дополнительные преимущества, удаляя мутантные факторы H, I, B, C3, антитела к фактору H и возмещая их функциональными белками. Многие авторы доказывают необходимость ежедневного плазмафереза длительное время . Ежедневная доза плазмы колеблется от 20 до 40 мл/кг, трансфузии производятся до достижения ремиссии

Эффективность ЗПП продемонстрирована в основном у пациентов с мутацией CFH. Однако, через несколько месяцев или лет возможно развитие вторичной резистентности к терапии СЗП, что ограничивает ее эффективность. Для пациентов с анти-CFH антителами ЗПП является методом выбора [6, 34]. Для предотвращения повышения уровня антител после ЗПП, сочетание последнего со стероидами и иммуносупрессивными препаратами представляется обоснованным

Противопоказанием к плазматерапии является ГУС, вызванный Streptococcus pneumonia, т.к. плазма взрослого человека содержит антитела против антигена Tromsen-Friedenreich, которые ухудшают течение процесса.

Большинство эпизодов аГУС связаны с инфекций, что обусловливает необходимость эрадикации хронических очагов аденоидной, тонзиллярной и зубной инфекции. У отдельных больных рецидивы отмечены после вакцинаций. Тем не менее, польза последних существенно превалирует над риском

Одной из важных клинических проблем является определение критериев прогноза заболевания. Так как около 50% выживших после острого периода в дальнейшем нуждаются в ЗПТ (заместительной почечной терапии) и трансплантации почки, то и прогноз в значительной степени определяется риском возврата аГУС после трансплантации

Перспективным направлением лечения аГУС является воздействие на систему комплемента, применение препарата, ингибирующего систему комплемента. К ним относится Экулизумаб (Солирис), человеческие моноклональные антитела к C5. Экулизумаб известен в терапии пароксизмальной ночной гемоглобинурии. Хотя при его употреблении несколько повышается риск инфекции Neisseria meningitides (необходимо до начала терапии привить ребенка), препарат в целом надежный и хорошо переносится. Результаты терапии отдельных больных аГУС с помощью экулизумаба весьма обнадеживающие. В целом, эти результаты свидетельствуют о перспективе экулизумаба, как препарата, являющегося новым стандартом лечения пациентов с аГУС. По результатам контролируемых клинических исследований, благодаря тому, что экулизумаб ингибирует комплемент-опосредованную ТМА, на терапии у 88% больных отсутствуют новые случаи развития ТМА, у 80% больных прекращается гемодиализ, 90% пациентов достигают нормализации гематологических показателей, у 100% больных прекращаются операции плазмообменов и нет новых больных, нуждающихся в гемодиализе. Более чем у половины больных отмечено улучшение функции почек, в частности у 47% достигнуто увеличение показателей СКФ≥15 мл/мин/1.73 м2. В связи с тем, что у больных аГУС неконтролируемая активация комплемента продолжается, риск внезапного развития осложнений сохраняется на протяжении всей жизни. Поэтому у таких пациентов рекомендуется постоянное проведение терапии экулизумабом

Проводятся клинические испытания растворимых форм ингибитора С3/С5-конвертазы, рецептора комплемента I (CRI) Для варианта с дефицитом HFI разрабатывается рекомбинантный HFI, а также возможность использования донорской плазмы, богатой HFI .











