. Это обусловлено нарушением регуляции активности Т-клеток или неспособнос…")
 нарушения системы иммунитета с дефектами одного или нескольких ее компонентов, а именно: клеточного или гуморального иммунитета, фагоцитоза, системы комплемента. В настоящее время идентифицированы многие дес…")
. нарушения иммунной…")

 гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов) Х-сцепленная агаммаглобулинемия (болезнь Брутона) Гипер-IgM синдром Х-сцепл…")
 тяжелая комбинированная иммунная недостаточность Х-сцепленная с полом аутосомно-рециссивная дефицит аденозиндезаминазы дефицит пуриннуклеозидфосфор…")
 циклическая нейтропения семейная доброкачественная нейтропения дефекты фагоцитарной функции хроническая гранулематозная болезнь …")
. I. Недостаточность гуморального звена иммунитета (системы В-лимфоцитов). 1. Агаммаглобулинемия, болезнь Брутона. 2. Дисгаммаглобулинемии: а) общая вариабельная гипогаммаглобули…")
. III. Комбинированные ИДС (тяжёлая комбинированная иммунологическая недостаточность - ТКИН). 1. Ретикулярная дисгенезия. 2. Наследственный алимфоцитоз, лимфоц…")
 синдром Чедиака-Хигаси; б) гипер IgE-синдром; 2. Нарушение процессов переваривания (киллинга) микробов: а) септический гранулемат…")




Презентация на тему: Понятие об иммунодефицитных состояниях и их классификация


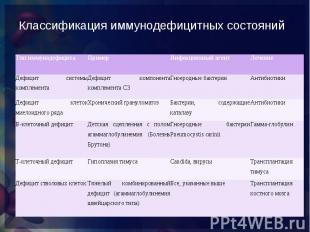
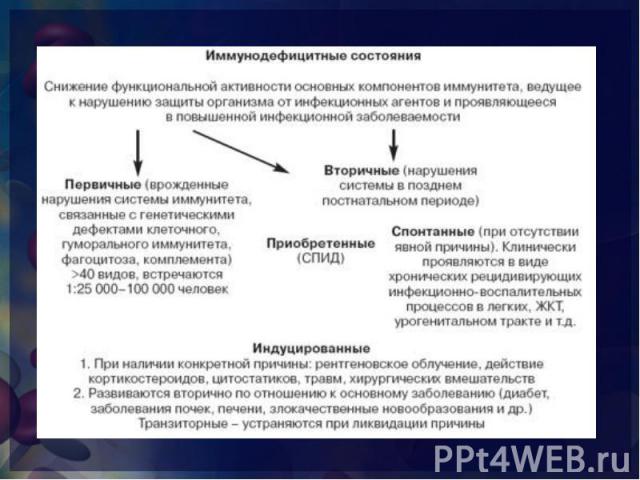
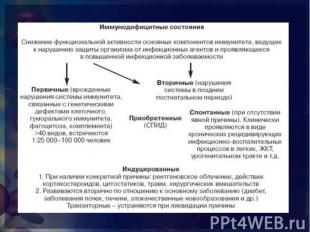
снижение функциональной активности основных компонентов системы иммунитета, ведущие к нарушению антигенного гомеостаза организма и прежде всего снижению способности организма защищаться от микробов и проявляющееся в повышенной инфекционной заболеваемости. снижение функциональной активности основных компонентов системы иммунитета, ведущие к нарушению антигенного гомеостаза организма и прежде всего снижению способности организма защищаться от микробов и проявляющееся в повышенной инфекционной заболеваемости. нарушения иммунологической реактивности, обусловленные выпадением одного или нескольких компонентов иммунного аппарата или тесно взаимодействующих с ним неспецифических факторов.

У людей, страдающих иммунодефицитом, отмечается высокая частота образования злокачественных опухолей и аутоантител (последнее может сопровождаться аутоиммунными заболеваниями). Это обусловлено нарушением регуляции активности Т-клеток или неспособностью организма справиться с основными вирусными заболеваниями. У людей, страдающих иммунодефицитом, отмечается высокая частота образования злокачественных опухолей и аутоантител (последнее может сопровождаться аутоиммунными заболеваниями). Это обусловлено нарушением регуляции активности Т-клеток или неспособностью организма справиться с основными вирусными заболеваниями.
 нарушения системы иммунитета с дефекта")
врожденные (генетические или эмбриопатии) нарушения системы иммунитета с дефектами одного или нескольких ее компонентов, а именно: клеточного или гуморального иммунитета, фагоцитоза, системы комплемента. В настоящее время идентифицированы многие десятки врожденных дефектов системы иммунитета, но очевидно, что действительное число ПИД значительно больше. Скрининг на ИД требует исследования у пациента состояния В-клеточных, Т-клеточных и комбинированных В- и Т-клеточных функций. Необходимо определение систем биологической амплификации (комплемента, цитокинов) и базисных эффекторных механизмов (фагоцитоза и воспалительного ответа) (СтИА, 2001). врожденные (генетические или эмбриопатии) нарушения системы иммунитета с дефектами одного или нескольких ее компонентов, а именно: клеточного или гуморального иммунитета, фагоцитоза, системы комплемента. В настоящее время идентифицированы многие десятки врожденных дефектов системы иммунитета, но очевидно, что действительное число ПИД значительно больше. Скрининг на ИД требует исследования у пациента состояния В-клеточных, Т-клеточных и комбинированных В- и Т-клеточных функций. Необходимо определение систем биологической амплификации (комплемента, цитокинов) и базисных эффекторных механизмов (фагоцитоза и воспалительного ответа) (СтИА, 2001).

нарушения иммунной системы, развивающиеся в постнеонатальном периоде у детей или у взрослых и не являющиеся результатом генетических дефектов. Среди ВтИД выделены три формы: приобретенная, индуцированная, спонтанная (СтИА, 2001,). нарушения иммунной системы, развивающиеся в постнеонатальном периоде у детей или у взрослых и не являющиеся результатом генетических дефектов. Среди ВтИД выделены три формы: приобретенная, индуцированная, спонтанная (СтИА, 2001,). Наиболее ярким примером приобретенной формы ВтИД является ВИЧ-инфекция с развитием синдрома приобретенного иммунодефицита (СПИД). Спонтанная форма ВтИД характеризуется отсутствием явной (очевидной) причины, вызвавшей нарушение иммунной реактивности у больного с чередой следующих друг за другом часто вяло текущих инфекционных заболеваний и отсутствием каких-либо отклонений в иммунном статусе (при современном уровне обследования). Спонтанная — это ВтИД с не выявленными причинами. Индуцированная форма ВтИД у детей наиболее часто вызвана дефицитами питания (в том числе и внутриутробными), инфекциями, среди которых особое место занимают внутриутробные, диарейным синдромом.

включают в себя группу заболеваний, клинически и иммунологически характеризующуюся дефектом как Т-, так и В-лимфоцитов. включают в себя группу заболеваний, клинически и иммунологически характеризующуюся дефектом как Т-, так и В-лимфоцитов. Комбинированные иммунодефициты, связанные с другими крупными дефектами включают заболевания, при которых иммунодефицит является одним из основных синдромов, но не единственным.

гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов) гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов) Х-сцепленная агаммаглобулинемия (болезнь Брутона) Гипер-IgM синдром Х-сцепленная аутосомно-рециссивная делеция генов тяжелых цепей иммуноглобулинов дефицит k-цепей селективный дефицит субклассов IgG с или без дефицита IgA дефицит антител с нормальным уровнем иммуноглобулинов общая вариабельная иммунная недостаточность дефицит IgA клеточные синдром Ди-Джоржи первичный дефицит CD4 клеток дефицит CD7 Т-клеток дефицит ИЛ-2 множественная недостаточность цитокинов дефект передачи сигнала

комбинированные: комбинированные: синдром Вискотта-Олдрича атаксия-телеангиоэктазия (синдром Луи-Бар) тяжелая комбинированная иммунная недостаточность Х-сцепленная с полом аутосомно-рециссивная дефицит аденозиндезаминазы дефицит пуриннуклеозидфосфорилазы дефицит молекул II класса МНС (синдром лысых лимфоцитов) ретикулярная дизгенезия дефицит CD3γ или CD3ε дефицит СD8 лимфоцитов недостаточность системы комплемента

дефекты фагоцитоза дефекты фагоцитоза наследственные нейтропении инфантильный летальный агранулоцитоз (болезнь Костмана) циклическая нейтропения семейная доброкачественная нейтропения дефекты фагоцитарной функции хроническая гранулематозная болезнь Х-сцепленная аутосомно-рециссивная дефицит адгезии лимфоцитов I типа дефицит адгезии лейкоцитов 2 типа дефицит глюкозо-6-дегидроегназы нейтрофилов дефицит миелопероксидазы дефицит вторичных гранул синдром Швахмана
. I. Недо")
I. Недостаточность гуморального звена иммунитета (системы В-лимфоцитов). I. Недостаточность гуморального звена иммунитета (системы В-лимфоцитов). 1. Агаммаглобулинемия, болезнь Брутона. 2. Дисгаммаглобулинемии: а) общая вариабельная гипогаммаглобулинемия; б) селективный дефицит IgA; в) дефицит иммуноглобулинов IgG и IgA с увеличением синтеза IgM - гипер IgM cиндром; г) дефицит подклассов IgG ( отсутствие IgG1, IgG2, IgG3, IgG4 с увеличением уровня IgM или без него); II. Недостаточность клеточных иммунных реакций (системы Т-лимфоцитов). 1. Лимфоцитарная дисгенезия (синдром Незелофа, французский тип ИДС). 2. Гипоплазия вилочковой железы и паращитовидных желёз (синдром Ди Джорджа).

III. Комбинированные ИДС (тяжёлая комбинированная иммунологическая недостаточность - ТКИН). III. Комбинированные ИДС (тяжёлая комбинированная иммунологическая недостаточность - ТКИН). 1. Ретикулярная дисгенезия. 2. Наследственный алимфоцитоз, лимфоцитофтиз (Швейцарский тип ИДС). 3. Синдром "голых" лимфоцитов. 4. ИДС с тимомой. 5. Синдром Вискотта-Олдрича. IV. Нарушения в системе интерлейкинов и кооперации клеток в иммунном ответе. V. ИДС при наследственных аномалиях обмена. 1. Недостаточность аденозиндеаминазы. 2. Недостаточность пуриннуклеотидфосфорилазы. VI. Недостаточность системы комплемента.

VII. Недостаточность фагоцитоза. VII. Недостаточность фагоцитоза. 1. Нарушение хемотаксиса, миграции и дегрануляции: а) синдром Чедиака-Хигаси; б) гипер IgE-синдром; 2. Нарушение процессов переваривания (киллинга) микробов: а) септический гранулематоз; б) липохромный гистиоцитоз; в) ферментопатии нейтрофильных гранулоцитов: дефицит миелопероксидазы, НАДН-оксидазы, глютатионпероксидазы, глюкозо-6-фосфатдегидрогеназы. 3. Дефекты опсонизации и поглощения: а) дефекты опсонизации; б) дефицит тафтсина; в) отсутствие мембранных гликопротеинов LAF-1, Gp150,95, Mac-1 и др.

I. Комбинированные ВИД. I. Комбинированные ВИД. 1. Общий лимфоцитопенический синдром. 2. Синдром поликлональной активации лимфоцитов. 3. Общий вариабельный иммунодефицит. 4. Синдром гиперплазии лимфоидной ткани (лимфоаденопатии, тимико-лимфатический синдром, тонзилогенный синдром). 5. Посттонзилэктомический синдром. II. Т-клеточные ВИД. 1. Т-лимфоцитопенический синдром. 2. Синдром Т-клеточного иммунорегуляторного дисбаланса. 3. Дефициты интерлейкинов, лимфокинов и их рецепторов.

III. В-клеточные ВИД. III. В-клеточные ВИД. 1. Общий вариабельный В-клеточный иммунодефицит. 2. Пангипогаммаглобулинемия. IV. Дефициты мононуклеарно-фагоцитарной системы. 1. Гранулоцитопении. 2. Дефициты рецепторов и адгезинов нейтрофилов. 3. Дефициты фагоцитоза. V. Дефициты молекул взаимодействия лейкоцитов. VI. Дефициты системы комплемента. 1. Синдром гипокомплементемии. 2. Дефициты отдельных факторов. VII. Дефициты нормальных киллеров. VIII. Дефициты неспецифических факторов иммунитета. IX. Метаболические ВИД.